Software
 |
 |
 |
| FragFit | MDsrv | MP:PD |
|---|---|---|
FragFit is an interactive web service for guided modeling of missing segments such as loops or hige regions into cryo-Electron Microscopy (cryo-EM) density maps. Segments of up to 3-35 residues length can be effectively modeled into maps of different resolution. Fragments are taken from a precalculated and frequently updated database extracted from structural coordinates deposited in the PDB. A fast hierarchical search algorithm selects up to 100 suitable fragments by using sequence similarity and geometrical fitting as evaluation criteria. The found fragments are then reranked by their cross-correlation to the user provided cryo-EM density map. |
MDsrv is a web-based tool developed to enhance collaborative research by providing non-experts with easy and quick on-line access to molecular dynamics (MD) simulations. As a member of the nglviewer family, MDsrv channels MD trajectories through a web server using a powerful web application (NGL viewer) to visualize them. |
Influence of structural dynamics and oligomerisation of ϒ-secretase substrates (APP, APLP) on their being processed to cell toxic species. Structural modelling of the modulation of the ϒ-sekretase section of APP by non-peptide active substances. Structural modelling of the modulation of fibril formation by peptide and non-peptide active substances. |
 |
 |
 |
| NGL | RHYTHM | SL2 |
NGL is an online viewer for proteins and other molecular structures. It is intended for users of the web application including a graphical interface and for developers who want to use NGL in their own projects. |
We work on the elucidation of the conformational space of GPCRs and its modulation by extracellular ligands and intracellular effector proteins by means of classical and umbrella sampling molecular dynamics simulations to address the issues of signaling specificity and biased signaling. For this reason we are computational modelling receptor G protein or arrestin interactions to elucidate the mechanism of signal transduction. |
SuperLooper2 is an interactive web service for the insertion of missing segments such as loops in proteins. Loop candidates are selected from a precalculated database containing ~700 million protein fragments with a residue length of 3-35. The fragments are extracted from structural coordinates deposited in the RSCB PDB. |
 |
 |
 |
ValiDen |
Voronoia |
mdciao |
The ValiDen webserver generates electron density maps. As a highlighting feature ValiDen provides the opportunity to convert the formats of electron microscopy maps (e.g. CCP4, X-PLOR, DSN6, BRIX) via the program MAPMAN in feasible map formats (e.g. MAP) for representation of EM-maps in e.g. COOT or PyMOL. |
Voronoia is a database storing data on atomic packing densities of RNA structures and complexes. Compared to other methods, that solely estimate van der Waals interactions i.e. by gain of solvent excluded surfaces, Voronoia4RNA allocates the free space between neighbouring atoms. It explicitly takes packing defects into consideration and is therefore an appropriate method to calculate van der Waals interactions and to estimate the underlying forces. |
mdciao is a Python module that provides quick, “one-shot” command-line tools and API methods to analyze MD data using residue-residue distances. It tries to automate as much as possible for non-experienced users while remaining highly customizable for advanced users. Ballesteros-Weinstein-Numbering (BW) and the Common G-alpha Numbering (CGN) consensus-nomenclature are included when possible. |
 |
||
| AlignMe | ProteinPrompt | SmoothT |
The AlignMe server creates alignments of membrane proteins and proteins that share hardly any sequence similarity but are related nonetheless. |
The ProteinPrompt server predicts protein-protein interactions (PPIs). Two implementations are available, one based on Random Forest, the other on Graph Neural Networks. Former scans the human proteom in around one minute, latter even faster. For a given query sequence a list of potential binders is returned. |
The SmoothT software allows the construction and visualization of low-energy pathways from an ensemble of molecular conformations, uploaded by the user. The pathways can be visualized as trajectories. |
 |
||
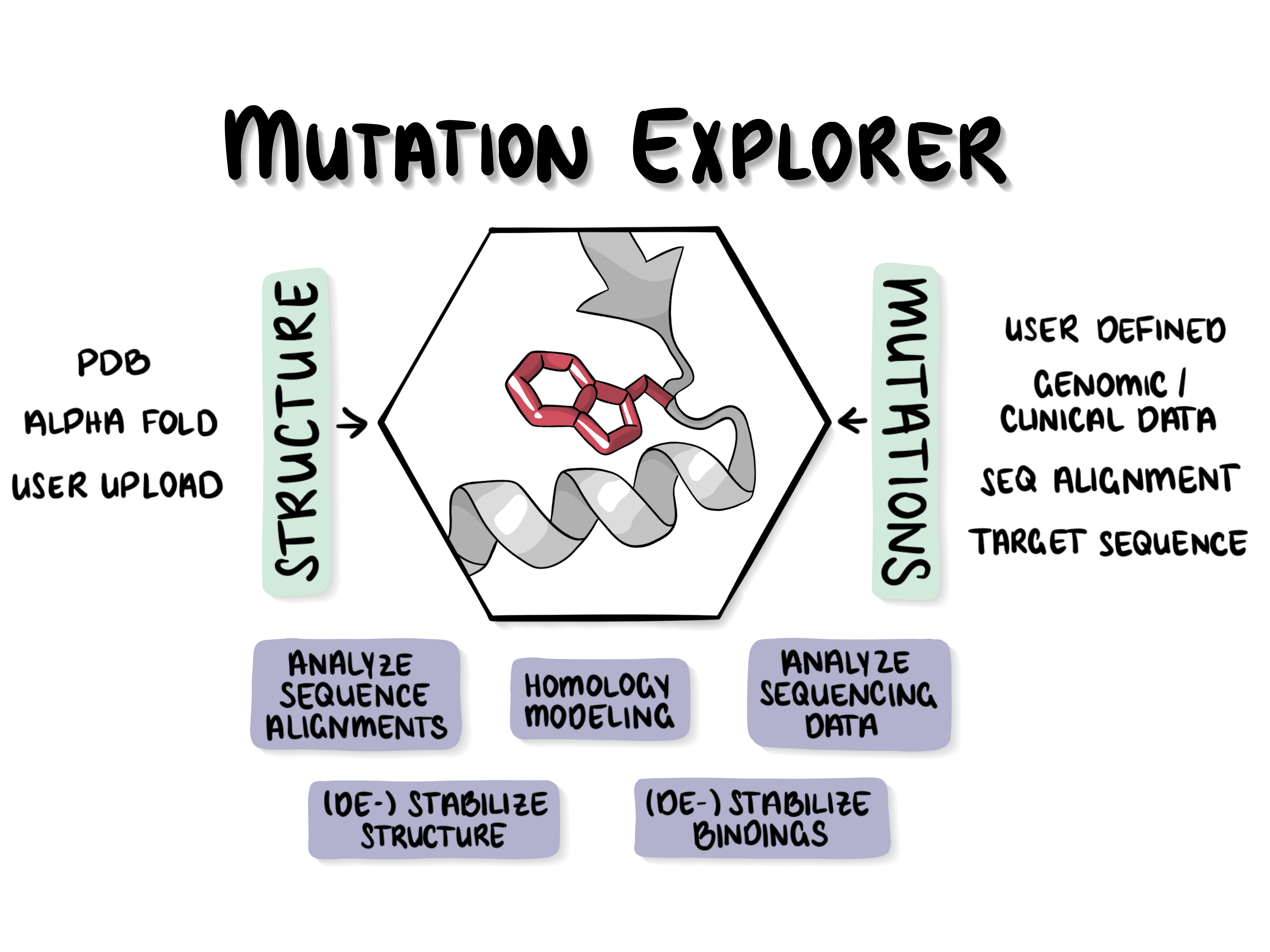
| The MutationExplorer | ||
The MutationExplorer webserver maps variants onto protein structures and allows users to study variation by inputting sequence changes. As the user enters variants, the 3D model evolves, and estimated changes in energy are highlighted. In addition to a basic per-residue input format, MutationExplorer can also upload an entire replacement sequence. Previously the purview of desktop applications, such an upload can back-mutate PDB structures to wildtype sequence in a single step. Structures are flexibly colorable, not only by energetic differences, but also by hydrophobicity, or sequence conservation, or other biochemical profiling. Another mutation source can be human single nucelotide polymorphisms (SNPs), genomic coordinates input in VCF format. |