Controlling Flareplots¶
The purpose of this notebook is to show the different ways in which a user can select which residues get shown or hidden in a flareplot, and how they can be broken down into different types of fragments to inform about the molecular topology.
We will try different ways of calling the method plot_freqs_as_flareplot, which is a class method of the object ContactGroup. Under the hood, the lower-level mdciao.flare.freqs2flare is at work, which is explained in this other notebook
[1]:
import mdciao
geom = mdciao.cli.pdb("3SN6")
fragments = mdciao.fragments.get_fragments(geom.top)
Checking https://files.rcsb.org/download/3SN6.pdb ...Please cite the following 3rd party publication:
* Crystal structure of the beta2 adrenergic receptor-Gs protein complex
Rasmussen, S.G. et al., Nature 2011
https://doi.org/10.1038/nature10361
Auto-detected fragments with method 'lig_resSeq+'
fragment 0 with 349 AAs THR9 ( 0) - LEU394 (348 ) (0) resSeq jumps
fragment 1 with 340 AAs GLN1 ( 349) - ASN340 (688 ) (1)
fragment 2 with 58 AAs ASN5 ( 689) - ARG62 (746 ) (2)
fragment 3 with 159 AAs ASN1002 ( 747) - ALA1160 (905 ) (3)
fragment 4 with 284 AAs GLU30 ( 906) - CYS341 (1189 ) (4) resSeq jumps
fragment 5 with 128 AAs GLN1 ( 1190) - SER128 (1317 ) (5)
fragment 6 with 1 AAs P0G1601 ( 1318) - P0G1601 (1318 ) (6)
These fragments do not exactly coincide with the chains (check this for more info), but they are useful for this example. The fragments are:
G-protein \(\alpha\) sub-unit
G-protein \(\beta\) sub-unit
G-protein \(\gamma\) sub-unit
Bacteriophage T4 lysozyme as N-terminus
\(\beta_2\) adrenergic receptor
VHH nanobody
Ligand P0G
Hence, we name them accordingly:
[2]:
fragment_names=['Galpha','Gbeta','Ggamma','T4L','B_2AR','NB','P0G']
frag_idxs_group variables, namely:frag_idxs_group_1=[0,1,2] : the G\(\alpha\), G\(\beta\), and G\(\gamma\) sub-unitsfrag_idxs_group_2=[4] : the B2AR receptorAnd we compute the interface without producing or saving any figures or files using the options figures=False and no_disk=True.
[3]:
GPCR = mdciao.nomenclature.LabelerGPCR("adrb2_human")
CGN = mdciao.nomenclature.LabelerCGN("3SN6")
intf = mdciao.cli.interface(geom,
frag_idxs_group_1=[0, 1, 2],
frag_idxs_group_2=[4],
no_disk=True,
figures=False,
fragment_names=fragment_names,
fragments=fragments,
GPCR_uniprot=GPCR, CGN_PDB=CGN,
accept_guess=True,
ctc_control=1.)
No local file ./adrb2_human.xlsx found, checking online in
https://gpcrdb.org/services/residues/extended/adrb2_human ...done!
Please cite the following reference to the GPCRdb:
* Kooistra et al, (2021) GPCRdb in 2021: Integrating GPCR sequence, structure and function
Nucleic Acids Research 49, D335--D343
https://doi.org/10.1093/nar/gkaa1080
For more information, call mdciao.nomenclature.references()
done without 404, continuing.
Using CGN-nomenclature, please cite
* Flock et al, (2015) Universal allosteric mechanism for G$\alpha$ activation by GPCRs
Nature 2015 524:7564 524, 173--179
https://doi.org/10.1038/nature14663
No local file ./CGN_3SN6.txt found, checking online in
https://www.mrc-lmb.cam.ac.uk/CGN/lookup_results/3SN6.txt ...done without 404, continuing.
No local PDB file for 3SN6 found in directory '.', checking online in
https://files.rcsb.org/download/3SN6.pdb ...found! Continuing normally
Will compute contact frequencies for trajectories:
<mdtraj.Trajectory with 1 frames, 10269 atoms, 1319 residues, and unitcells>
with a stride of 1 frames
Using method 'user input by residue array or range' these fragments were found
fragment 0 with 349 AAs THR9 ( 0) - LEU394 (348 ) (0) resSeq jumps
fragment 1 with 340 AAs GLN1 ( 349) - ASN340 (688 ) (1)
fragment 2 with 58 AAs ASN5 ( 689) - ARG62 (746 ) (2)
fragment 3 with 159 AAs ASN1002 ( 747) - ALA1160 (905 ) (3)
fragment 4 with 284 AAs GLU30 ( 906) - CYS341 (1189 ) (4) resSeq jumps
fragment 5 with 128 AAs GLN1 ( 1190) - SER128 (1317 ) (5)
fragment 6 with 1 AAs P0G1601 ( 1318) - P0G1601 (1318 ) (6)
GPCR-labels align best with fragments: [4] (first-last: GLU30-CYS341).
These are the GPCR fragments mapped onto your topology:
TM1 with 32 AAs GLU30@1.29x29 ( 906) - PHE61@1.60x60 (937 ) (TM1)
ICL1 with 4 AAs GLU62@12.48x48 ( 938) - GLN65@12.51x51 (941 ) (ICL1)
TM2 with 32 AAs THR66@2.37x37 ( 942) - LYS97@2.68x67 (973 ) (TM2)
ECL1 with 4 AAs THR98@23.49x49 ( 974) - PHE101@23.52x52 (977 ) (ECL1)
TM3 with 36 AAs GLY102@3.21x21 ( 978) - SER137@3.56x56 (1013 ) (TM3)
ICL2 with 8 AAs PRO138@34.50x50 ( 1014) - LEU145@34.57x57 (1021 ) (ICL2)
TM4 with 27 AAs THR146@4.38x38 ( 1022) - HIS172@4.64x64 (1048 ) (TM4)
ECL2 with 20 AAs TRP173 ( 1049) - THR195 (1068 ) (ECL2) resSeq jumps
TM5 with 42 AAs ASN196@5.35x36 ( 1069) - GLU237@5.76x76 (1110 ) (TM5)
ICL3 with 2 AAs GLY238 ( 1111) - ARG239 (1112 ) (ICL3)
TM6 with 35 AAs CYS265@6.27x27 ( 1113) - GLN299@6.61x61 (1147 ) (TM6)
ECL3 with 4 AAs ASP300 ( 1148) - ILE303 (1151 ) (ECL3)
TM7 with 25 AAs ARG304@7.31x30 ( 1152) - ARG328@7.55x55 (1176 ) (TM7)
H8 with 13 AAs SER329@8.47x47 ( 1177) - CYS341@8.59x59 (1189 ) (H8)
CGN-labels align best with fragments: [0] (first-last: THR9-LEU394).
These are the CGN fragments mapped onto your topology:
G.HN with 28 AAs THR9@G.HN.26 ( 0) - VAL36@G.HN.53 (27 ) (G.HN)
G.hns1 with 3 AAs TYR37@G.hns1.1 ( 28) - ALA39@G.hns1.3 (30 ) (G.hns1)
G.S1 with 7 AAs THR40@G.S1.1 ( 31) - LEU46@G.S1.7 (37 ) (G.S1)
G.s1h1 with 6 AAs GLY47@G.s1h1.1 ( 38) - GLY52@G.s1h1.6 (43 ) (G.s1h1)
G.H1 with 7 AAs LYS53@G.H1.1 ( 44) - GLN59@G.H1.7 (50 ) (G.H1)
H.HA with 26 AAs LYS88@H.HA.4 ( 51) - LEU113@H.HA.29 (76 ) (H.HA)
H.hahb with 9 AAs VAL114@H.hahb.1 ( 77) - PRO122@H.hahb.9 (85 ) (H.hahb)
H.HB with 14 AAs GLU123@H.HB.1 ( 86) - ASN136@H.HB.14 (99 ) (H.HB)
H.hbhc with 7 AAs VAL137@H.hbhc.1 ( 100) - PRO143@H.hbhc.15 (106 ) (H.hbhc)
H.HC with 12 AAs PRO144@H.HC.1 ( 107) - GLU155@H.HC.12 (118 ) (H.HC)
H.hchd with 1 AAs ASP156@H.hchd.1 ( 119) - ASP156@H.hchd.1 (119 ) (H.hchd)
H.HD with 12 AAs GLU157@H.HD.1 ( 120) - GLU168@H.HD.12 (131 ) (H.HD)
H.hdhe with 5 AAs TYR169@H.hdhe.1 ( 132) - ASP173@H.hdhe.5 (136 ) (H.hdhe)
H.HE with 13 AAs CYS174@H.HE.1 ( 137) - LYS186@H.HE.13 (149 ) (H.HE)
H.hehf with 7 AAs GLN187@H.hehf.1 ( 150) - SER193@H.hehf.7 (156 ) (H.hehf)
H.HF with 6 AAs ASP194@H.HF.1 ( 157) - ARG199@H.HF.6 (162 ) (H.HF)
G.hfs2 with 5 AAs CYS200@G.hfs2.1 ( 163) - GLY206@G.hfs2.7 (167 ) (G.hfs2) resSeq jumps
G.S2 with 8 AAs ILE207@G.S2.1 ( 168) - VAL214@G.S2.8 (175 ) (G.S2)
G.s2s3 with 2 AAs ASP215@G.s2s3.1 ( 176) - LYS216@G.s2s3.2 (177 ) (G.s2s3)
G.S3 with 8 AAs VAL217@G.S3.1 ( 178) - VAL224@G.S3.8 (185 ) (G.S3)
G.s3h2 with 3 AAs GLY225@G.s3h2.1 ( 186) - GLN227@G.s3h2.3 (188 ) (G.s3h2)
G.H2 with 10 AAs ARG228@G.H2.1 ( 189) - CYS237@G.H2.10 (198 ) (G.H2)
G.h2s4 with 5 AAs PHE238@G.h2s4.1 ( 199) - THR242@G.h2s4.5 (203 ) (G.h2s4)
G.S4 with 7 AAs ALA243@G.S4.1 ( 204) - ALA249@G.S4.7 (210 ) (G.S4)
G.s4h3 with 8 AAs SER250@G.s4h3.1 ( 211) - ASN264@G.s4h3.15 (218 ) (G.s4h3) resSeq jumps
G.H3 with 18 AAs ARG265@G.H3.1 ( 219) - LEU282@G.H3.18 (236 ) (G.H3)
G.h3s5 with 3 AAs ARG283@G.h3s5.1 ( 237) - ILE285@G.h3s5.3 (239 ) (G.h3s5)
G.S5 with 7 AAs SER286@G.S5.1 ( 240) - ASN292@G.S5.7 (246 ) (G.S5)
G.s5hg with 1 AAs LYS293@G.s5hg.1 ( 247) - LYS293@G.s5hg.1 (247 ) (G.s5hg)
G.HG with 17 AAs GLN294@G.HG.1 ( 248) - ASP310@G.HG.17 (264 ) (G.HG)
G.hgh4 with 10 AAs TYR311@G.hgh4.1 ( 265) - THR320@G.hgh4.10 (274 ) (G.hgh4)
G.H4 with 27 AAs PRO321@G.H4.1 ( 275) - ARG347@G.H4.27 (301 ) (G.H4)
G.h4s6 with 11 AAs ILE348@G.h4s6.1 ( 302) - TYR358@G.h4s6.20 (312 ) (G.h4s6)
G.S6 with 5 AAs CYS359@G.S6.1 ( 313) - PHE363@G.S6.5 (317 ) (G.S6)
G.s6h5 with 5 AAs THR364@G.s6h5.1 ( 318) - ASP368@G.s6h5.5 (322 ) (G.s6h5)
G.H5 with 26 AAs THR369@G.H5.1 ( 323) - LEU394@G.H5.26 (348 ) (G.H5)
Computing distances in the interface between fragments
0, 1, 2
and
4
The interface is restricted to the residues within 35.0 Angstrom of each other in the reference topology.
Computing interface...done!
From 212148 potential group_1-group_2 distances, the interface was reduced to only 42693 potential contacts.
If this number is still too high (i.e. the computation is too slow) consider using a smaller interface cutoff
0%| | 0/1 [00:00<?, ?it/s]
Streaming over trajectory object nr. 0 ( 1 frames, 1 with stride 1) in chunks of 10000 frames. Now at chunk nr 0, frames so far 1
100%|███████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████| 1/1 [00:02<00:00, 2.46s/it]
These 20 contacts capture 20.00 (~100%) of the total frequency 20.00 (over 42693 input contacts)
As orientation value, the first 18 ctcs already capture 90.0% of 20.00.
The 18-th contact has a frequency of 1.00
freq label residue idxs sum
0 1.0 R385@G.H5.17 - I233@5.72x72 339 1106 1.0
1 1.0 H387@G.H5.19 - A134@3.53x53 341 1010 2.0
2 1.0 T350@G.h4s6.3 - R239@ICL3 304 1112 3.0
3 1.0 Q384@G.H5.16 - P138@34.50x50 338 1014 4.0
4 1.0 D381@G.H5.13 - K232@5.71x71 335 1105 5.0
5 1.0 L393@G.H5.25 - A271@6.33x33 347 1119 6.0
6 1.0 F376@G.H5.8 - F139@34.51x51 330 1015 7.0
7 1.0 Q384@G.H5.16 - I135@3.54x54 338 1011 8.0
8 1.0 Q384@G.H5.16 - E225@5.64x64 338 1098 9.0
9 1.0 R385@G.H5.17 - Q229@5.68x68 339 1102 10.0
10 1.0 L393@G.H5.25 - T274@6.36x36 347 1122 11.0
11 1.0 Q384@G.H5.16 - Q229@5.68x68 338 1102 12.0
12 1.0 P122@H.hahb.9 - Q337@8.55x55 85 1185 13.0
13 1.0 Q125@H.HB.3 - Q337@8.55x55 88 1185 14.0
14 1.0 R380@G.H5.12 - T136@3.55x55 334 1012 15.0
15 1.0 R380@G.H5.12 - F139@34.51x51 334 1015 16.0
16 1.0 E392@G.H5.24 - T274@6.36x36 346 1122 17.0
17 1.0 I383@G.H5.15 - P138@34.50x50 337 1014 18.0
18 1.0 Y358@G.h4s6.20 - I233@5.72x72 312 1106 19.0
19 1.0 E392@G.H5.24 - K270@6.32x32 346 1118 20.0
label freq
0 Q384@G.H5.16 4.0
1 R385@G.H5.17 2.0
2 L393@G.H5.25 2.0
3 R380@G.H5.12 2.0
4 E392@G.H5.24 2.0
5 H387@G.H5.19 1.0
6 T350@G.h4s6.3 1.0
7 D381@G.H5.13 1.0
8 F376@G.H5.8 1.0
9 P122@H.hahb.9 1.0
10 Q125@H.HB.3 1.0
11 I383@G.H5.15 1.0
12 Y358@G.h4s6.20 1.0
label freq
0 I233@5.72x72 2.0
1 P138@34.50x50 2.0
2 F139@34.51x51 2.0
3 Q229@5.68x68 2.0
4 T274@6.36x36 2.0
5 Q337@8.55x55 2.0
6 A134@3.53x53 1.0
7 R239@ICL3 1.0
8 K232@5.71x71 1.0
9 A271@6.33x33 1.0
10 I135@3.54x54 1.0
11 E225@5.64x64 1.0
12 T136@3.55x55 1.0
13 K270@6.32x32 1.0
The scheme option in plot_freqs_as_flareplot¶
The scheme option controls which residues get shown in the flareplot, and how they get split into fragments. This choice can vary between all residues and only those strictly necessary.
Depending what exactly the user wants to highlight, there’s a tradeoff between the number of residues shown and how legible the plot remains. On the one hand, the more residues you show, the more possibilities you have to display the sequence and overall topology of the system with its fragments, e.g. receptor and G-protein and perhaps even sub-domain fragmentation, e.g. breaking the receptor into helices and loops. On the other hand, more residues means smaller fontsizes and possibly a lot of seemingly unused blank space.
Let’s go over the main options for scheme. Since we’re leaving some out, check the docs to see all possibilities.
scheme=='all'¶
From the docs:
How to decide which residues to plot
* 'all'
plot as many residues as possible. E.g.,
if a :obj:`self.topology` is present,
plot all its residues. This can be modified
with :obj:`fragments`, see above.
[4]:
ifig, iax = intf.plot_freqs_as_flareplot(3.5, scheme="all");
ifig.savefig("flare.all.pdf")
Drawing this many dots (1319 residues + 3 padding spaces) in a panel 10.0 inches wide/high
forces too small dotsizes and fontsizes. If crowding effects occur, either reduce the
number of residues or increase the panel size
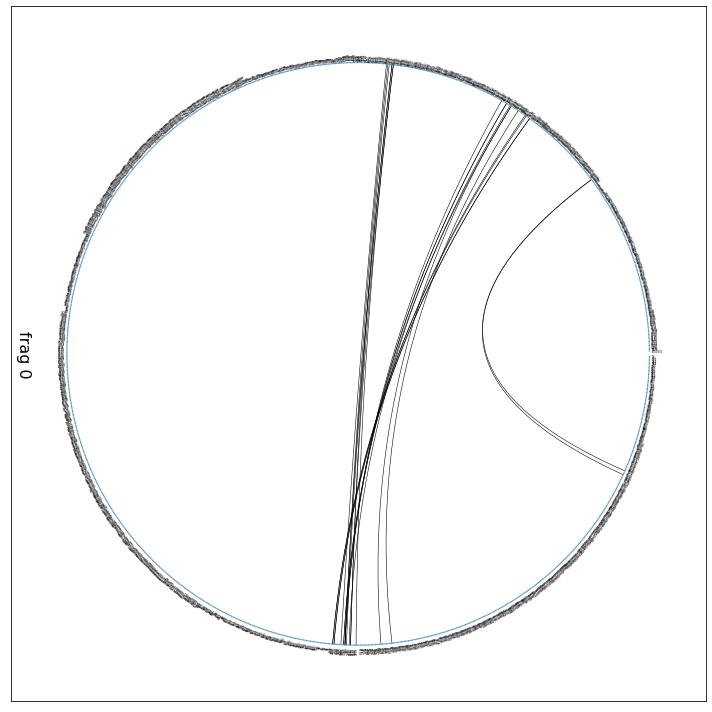
Definitively hard to see anything in the notebook. However, open flare.all.pdf externally and zoom in. You’ll see that all information is there. Let’s include the fragment information from above, which was:
fragments = mdciao.fragments.get_fragments(geom.top)
fragment_names=['Galpha','Gbeta','Ggamma','T4L','B_2AR','NB','P0G']
[5]:
ifig, iax = intf.plot_freqs_as_flareplot(3.5,
scheme="all",
fragments=fragments,
fragment_names=fragment_names)
ifig.savefig("flare.all.w_fragments.pdf")
Drawing this many dots (1319 residues + 9 padding spaces) in a panel 10.0 inches wide/high
forces too small dotsizes and fontsizes. If crowding effects occur, either reduce the
number of residues or increase the panel size
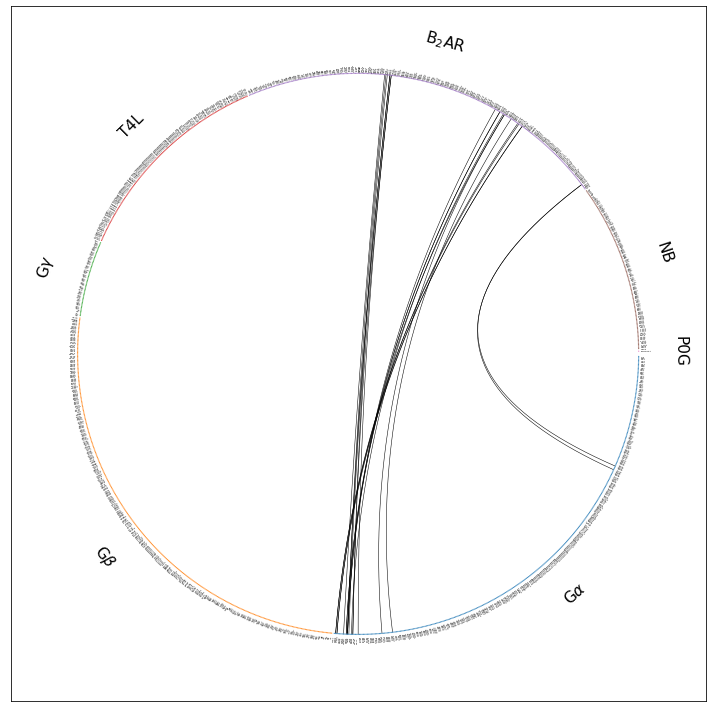
Still barely legible in the notebook, but zoom into flare.all.w_fragments.pdf and you’ll notice a couple of things:
The residue dots are now grouped into fragments
The fragments are named
The fragments (their residues’ dots) are color-coded, e.g. G\(\alpha\) is blue, receptor is B\(_2\)AR is violet
Unused Space : scheme='interface'¶
While it might be usefull to plot all residues and fragments of the topology, most of the flareplot is unused. E.g., we know for sure that the 4TL and the NB won’t get any contacts, because they simply were not considered when defining the interface, as we did above:
frag_idxs_group_1=[0,1,2]: the G\(\alpha\), G\(\beta\), and G\(\gamma\) sub-unitsfrag_idxs_group_2=[4]: the B2AR receptor
So, we can hide 4TL and the NB by using the scheme=interface option, which will automatically hide fragments that weren’t even considered in the interface definition. This is possible because the residues defining these fragments are stored internally in the `intf.interface_fragments <>`__ object, and get re-used here.
From the docs:
* 'interface':
use only the fragments in
:obj:`self.interface_fragments`. Will
only work if self.is_interface is True
[6]:
intf.plot_freqs_as_flareplot(3.5,
scheme="interface",
fragments=fragments,
fragment_names=fragment_names,
);
ifig.savefig("flare.interface.w_fragments.pdf")
Drawing this many dots (1031 residues + 6 padding spaces) in a panel 10.0 inches wide/high
forces too small dotsizes and fontsizes. If crowding effects occur, either reduce the
number of residues or increase the panel size
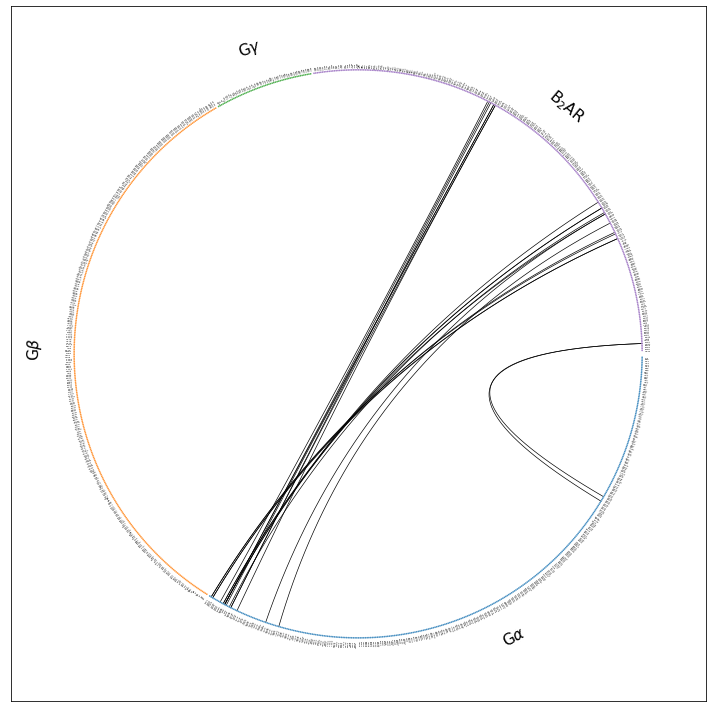
Now, we only have the fragments that were interesting to us to begin with. Still, a lot of unused space, because some of the fragments we included in the interface definition (G\(\beta\) and G\(\gamma\)), as potential interface partners, don’t have any contacts with the B2AR (at 3.5 Angstrom) in the 3SN6 structure. So, we hide them in the next paragraph.
Unused Space: scheme=interface_sparse¶
From the docs:
* 'interface_sparse':
like 'interface', but using the input :obj:`fragments`
to break self.interface_fragments (which are only two,
by definition) further down into other fragments.
Of these, show only the ones where at least one residue
participates in the interface. If :obj:`fragments` is
None, `scheme='interface'` and `scheme='interface_sparse'`
are the same thing.
[7]:
ifig, iax = intf.plot_freqs_as_flareplot(3.5,
scheme="interface_sparse",
fragments=fragments,
fragment_names=fragment_names,
SS=True,
);
ifig.savefig("flare.interface_sparse.w_fragments.pdf")
Drawing this many dots (633 residues + 4 padding spaces) in a panel 10.0 inches wide/high
forces too small dotsizes and fontsizes. If crowding effects occur, either reduce the
number of residues or increase the panel size
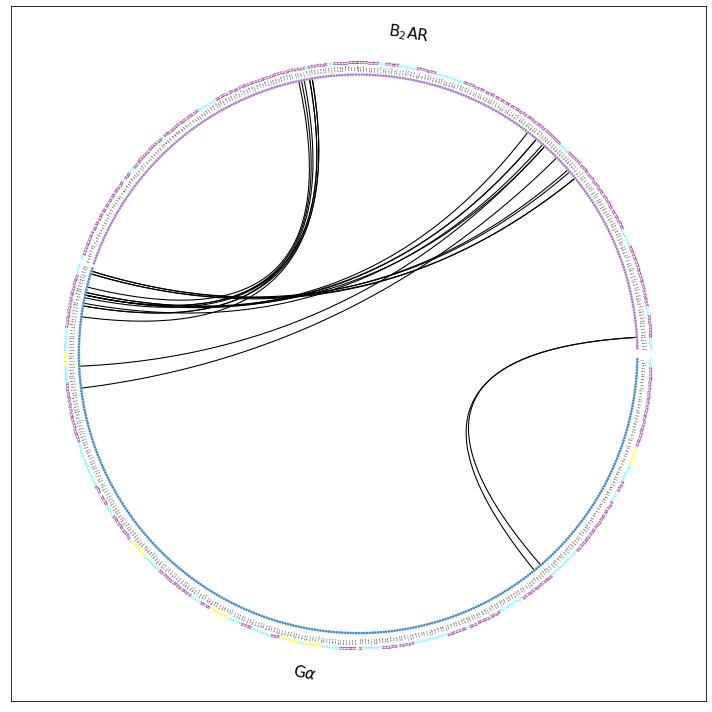
Note that the residues are now annotated with their secondary structure (SS=True) as an extra color-coded ring of letters:
\(\alpha\)-helix is purple H
\(\beta\)-sheet is yellow B
random coil is cyan C
Now, we can e.g. identify the helices and loops. E.g. we see the CT of the G\(\alpha\) (around 9 o’clock in the plot) is responsible for most contacts. It is mostly helical, except for the last, terminal residues. In the B2AR, we can identify helical domains and the loops connecting them. If we walk the the B2AR-fragment counter-clockwise (starting at around 3 o’clock), we see the last few CT residues as coil, then H8, then the H8-TM7 coil, then H7, then a loop, then TM6, then a loop, and then TM5.
Using the Blank Space¶
The next step is either to get rid of the unused, blank space (see further down) or to use it for something. In the next paragraph, we use it to inform about sub-fragments of the topology. In our case, we do this by using
Consensus Labels¶
We now incorporate consensus labels for the receptor and the G-protein into the flareplot. In mdciao, consensus labels are dealt with using the LabelerConsensus objects. These were created at the very top of the notebook with :
GPCR = mdciao.nomenclature.LabelerGPCR("adrb2_human")
CGN = mdciao.nomenclature.LabelerCGN("3SN6")
These objects are very versatile and can be reused with multiple topologies or multiple function calls. Because of that flexibility, there’s two ways in which we can use them in the flareplot, via the consensus_maps argument, with different results. From the docs:
consensus_maps : list, default is None
The items of this list are either:
* indexables containing the consensus
labels (strings) themselves. They
need to be "gettable" by residue index, i.e.
dict, list or array. Typically, one
generates these maps by using the top2labels
method of the LabelerConsensus object
* :obj:`LabelerConsensus`-objects
When these objects are passed, their
top2labels and top2fragments methods are
called on-the-fly, generating not only
the consensus labels but also the consensus
fragments (i.e. subdomains) to further fragment
the topology into sub-domains, like TM6 or G.H5.
If :obj:`fragments` are parsed, they will be
made compatible with the consensus fragments.
If you want the consensus labels but not
the sub-fragmentation, simply use the first option.
We will use second option, which will automaticall a) annotate residues and b) fragment the topology (the dots) further into sub-domains corresponding to the consensus sub-domains.
[8]:
ifig, iax = intf.plot_freqs_as_flareplot(3.5,
scheme="interface_sparse",
fragments=fragments,
fragment_names=fragment_names,
consensus_maps=[GPCR, CGN],
SS=True)
ifig.savefig("flare.interface_sparse.w_fragments.w_consensus.pdf")
Drawing this many dots (633 residues + 52 padding spaces) in a panel 10.0 inches wide/high
forces too small dotsizes and fontsizes. If crowding effects occur, either reduce the
number of residues or increase the panel size
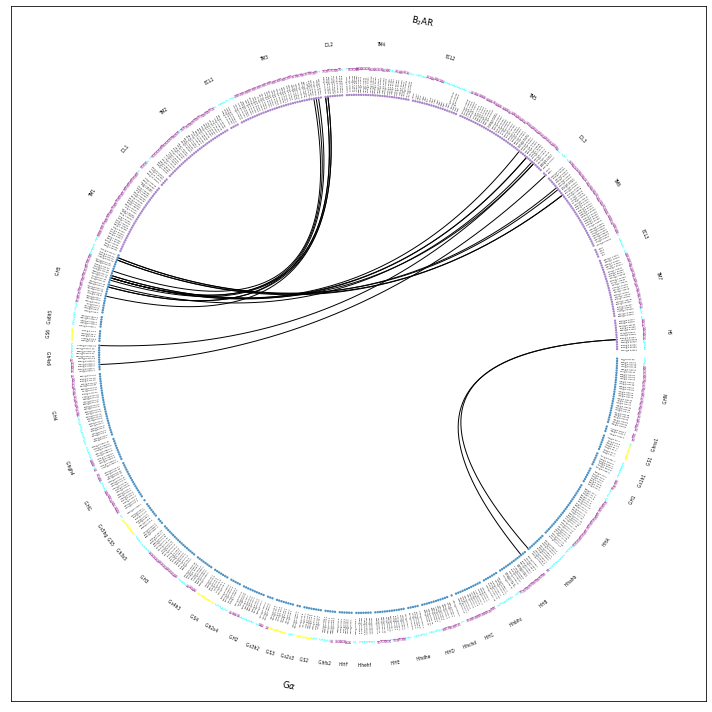
Same amount of blank space, but now at least we get to see how the consensus descriptors are used to fragment the G\(\alpha\) and the B\(_2\)AR further. Also, all residues are annotated with their consensus labels, e.g. L393@G.H5.25 and A271@6.33. Legibility remains an issue in the notebook, but check flare.interface_sparse.w_fragments.w_consensus.pdf and you’ll see all annotations clearly there.
Unused Sub-fragments: scheme='consensus_sparse'¶
We apply the same sparse logic as before, but to the consensus labels, i.e., only show consensus fragments that were part of the interface. From the docs:
* 'consensus_sparse':
like 'interface_sparse', but
leaving out sub-domains not participating
in the interface with any contacts.For this,
the :obj:`consensus_maps` need to
be actual :obj:`LabelerConsensus`-objects
[9]:
ifig, iax = intf.plot_freqs_as_flareplot(3.5,
scheme="consensus_sparse",
fragments=fragments,
fragment_names=fragment_names,
consensus_maps=[GPCR, CGN],
SS=True,
);
ifig.savefig("flare.consensus.w_fragments.w_consensus.pdf")
Drawing this many dots (196 residues + 12 padding spaces) in a panel 10.0 inches wide/high
forces too small dotsizes and fontsizes. If crowding effects occur, either reduce the
number of residues or increase the panel size
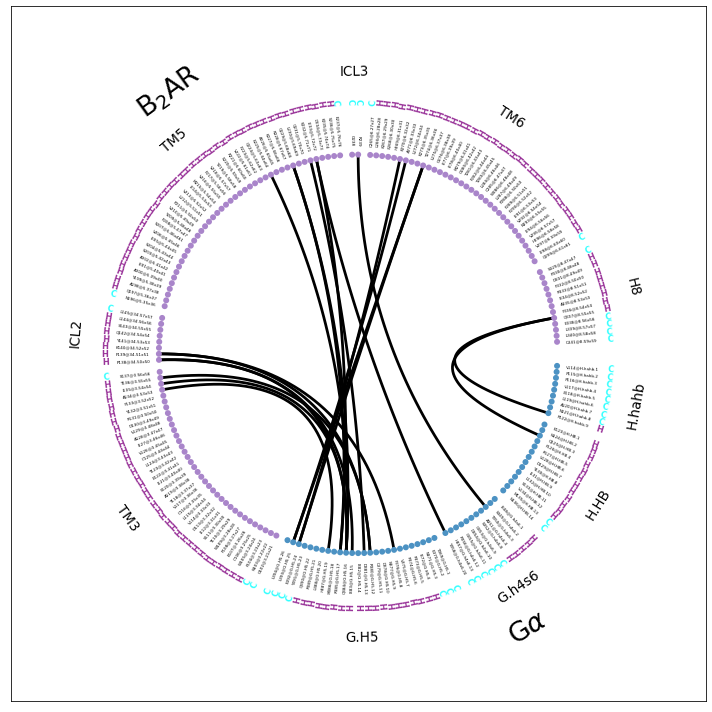
This seems to be a very good compromise for the representation. On the one hand, we’re leaving out a lot of unused sub-domain fragments, e.g. the extracellular loops (and some helices) of the receptor and the many parts of the G\(\alpha\) far away from the interface (e.g. the entire RAS domain). On the other hand, the topology is still somewhat represented, with full helices and loops clearly noted and visually separated. Of course, there still is some distortion and sequence jumps (e.g ICL2 to TM5 or G.h4s6 to G.H5) but these are easy to spot and are informative themselves: if TM4 and ECL2 aren’t shown, they are not participating in the computed interface.
Unused Residues: scheme='residues'¶
We can even get rid of all residues not involved in the interface. From the docs:
* 'residues':
plot only the residues present in self.res_idxs_pairs
If the interface is plotted using the same ctc_cutoff_Ang, these residues will be those listed in the initial output, on top of the notebook:
freq label residue idxs sum
0 1.0 R385@G.H5.17 - I233@5.72 339 1106 1.0
1 1.0 H387@G.H5.19 - A134@3.53 341 1010 2.0
2 1.0 T350@G.h4s6.3 - R239@ICL3 304 1112 3.0
3 1.0 Q384@G.H5.16 - P138@34.50 338 1014 4.0
4 1.0 D381@G.H5.13 - K232@5.71 335 1105 5.0
5 1.0 L393@G.H5.25 - A271@6.33 347 1119 6.0
6 1.0 F376@G.H5.8 - F139@34.51 330 1015 7.0
7 1.0 Q384@G.H5.16 - I135@3.54 338 1011 8.0
8 1.0 Q384@G.H5.16 - E225@5.64 338 1098 9.0
9 1.0 R385@G.H5.17 - Q229@5.68 339 1102 10.0
10 1.0 L393@G.H5.25 - T274@6.36 347 1122 11.0
11 1.0 Q384@G.H5.16 - Q229@5.68 338 1102 12.0
12 1.0 P122@H.hahb.9 - Q337@8.55 85 1185 13.0
13 1.0 Q125@H.HB.3 - Q337@8.55 88 1185 14.0
14 1.0 R380@G.H5.12 - T136@3.55 334 1012 15.0
15 1.0 R380@G.H5.12 - F139@34.51 334 1015 16.0
16 1.0 E392@G.H5.24 - T274@6.36 346 1122 17.0
17 1.0 I383@G.H5.15 - P138@34.50 337 1014 18.0
18 1.0 Y358@G.h4s6.20 - I233@5.72 312 1106 19.0
19 1.0 E392@G.H5.24 - K270@6.32 346 1118 20.0
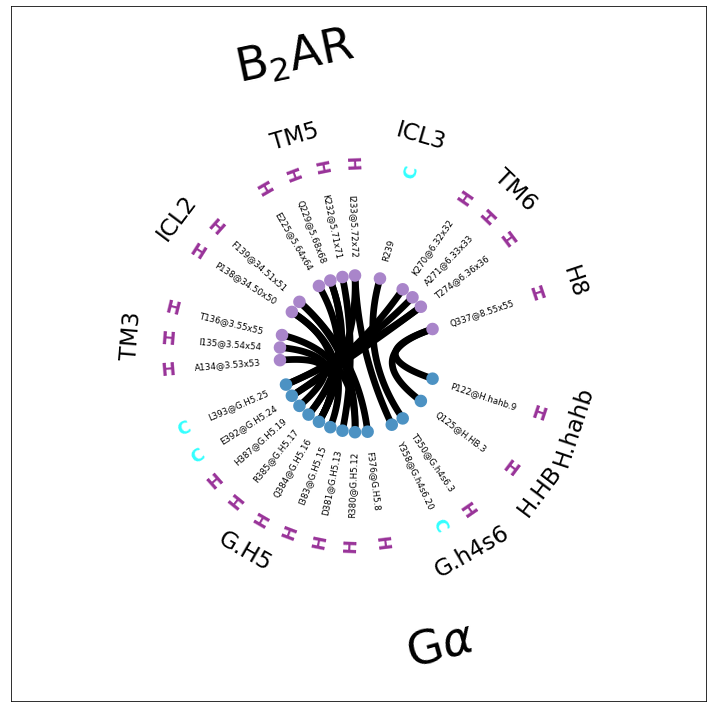
Depending on the usecase, this can be helpful or not, since most of the topology information is now lost: subfragments are kept but they there’s sequence-jumps within them (e.g. TM5, TM6 and G.H5).
[10]:
ifig, iax = intf.plot_freqs_as_flareplot(3.5,
scheme="residues",
fragments=fragments,
fragment_names=fragment_names,
consensus_maps=[GPCR, CGN],
SS=True,
);
ifig.savefig("flare.residues.w_fragments.w_consensus.pdf")
Coarse-Graning Flareplots: Chord Diagrams¶
Finally, we can choose to coarse-grain the flareplot into a chord-diagram. For this, per-residue contact frequencies are aggregated by fragment, highlighting a fragment’s participation in the interface, rather than each residue’s.
[11]:
ifig, iax = intf.plot_freqs_as_flareplot(3.5,
scheme="all",
fragments=fragments,
fragment_names=fragment_names,
consensus_maps=[GPCR, CGN],
SS=True,
coarse_grain=True,
);
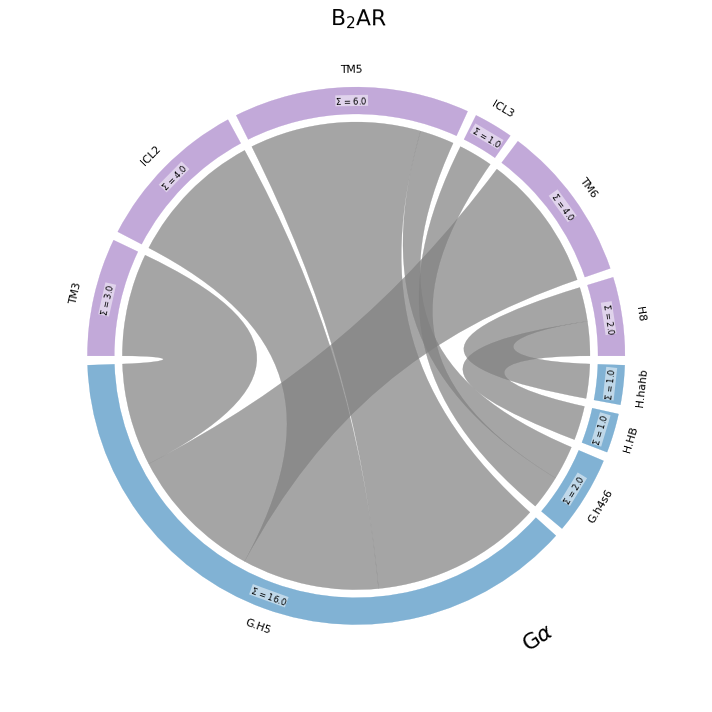
Here, the per-fragment contact frequencies are not represented as curve opacities, but as arc lengths. The the more contacts a fragment has, longer the arc used to represent it. In this case, it’s immediately clear that the G.H5 sub-domain of the G\(\alpha\) (also called G\(\alpha_5\)), with 16 contacts, clearly dominates the G-protein’s participation in the interface with the B2AR.
Some observations:
The size of the fragment doesn’t necessarily play a role here: only its participation in the interface. E.g. TM3 is clearly a larger fragment than ICL2 (check this plot), but they appear similar in size because their participation in the interface is similar.
The keyword
schemeloses meaning here: chord-diagrams are always sparse, i.e. zero-length fragments are never shown. As you see, thescheme='all'isn’t having any effect.Under the hood, mdciao.flare.freqs2chord is being, called, which itself wraps around the mpl_chord_diagram package.
The data being represented is the coarse-grained frequency matrix, which is calculated using mdciao.contacts.ContactGroup.frequency_as_contact_matrix_CG
[12]:
intf.frequency_as_contact_matrix_CG(3.5,
fragments=fragments,
fragment_names=fragment_names,
consensus_labelers=[GPCR, CGN],
interface=True).replace(0, "")
[12]:
| TM3 | ICL2 | TM5 | ICL3 | TM6 | H8 | |
|---|---|---|---|---|---|---|
| H.hahb | 1.0 | |||||
| H.HB | 1.0 | |||||
| G.h4s6 | 1.0 | 1.0 | ||||
| G.H5 | 3.0 | 4.0 | 5.0 | 4.0 |