Below you will find a very simple example of how to use mdciao from the command-line. Keep scrolling to the Highlights for more elaborate CLI-examples or jump to the Jupyter Notebook for a minimal API walkthrough. For real usecase and some context on definitions etc, go to the Jupyter Notebook Gallery.
Data and 3D visualization
The simulation data for generating these examples was kindly provided by Dr. H. Batebi. It can be 3D-visualized interactively here while checking out the examples. You can also download mdciao_example.zip and follow along.
Copying Terminal Commands
All code snippets can be copied and pasted directly into your terminal using the copy icon in the upper right of the snippet’s frame:
>>> Clickthebuttonatendoftheframetocopythistext;)
This basic command:
mdc_neighborhoods.pytop.pdbtraj.xtc--residuesL394-nf#nf: don't use fragments
will print the following to the terminal (some headers have been left out):
Fig. 1 [neighborhood.overall@4.5_Ang.pdf] Using 4.5 Å as distance cutoff, the most frequent neighbors of LEU394, the C-terminal residue in the \(\alpha_5\) helix of the Gs-protein, are shown. \(\Sigma\) is the sum over frequencies and represents the average number of neighbors of LEU394. The simulation started from the 3SN6 structure (beta2 adrenergic receptor-Gs protein complex, no antibody). The simulation itself can be seen interactively in 3D here.
Annotated figures with the timetraces of the above distances are also produced automatically:
Fig. 2 [neighborhood.LEU394.time_trace@4.5_Ang.pdf] Time-traces of the residue-residue distances used for the frequencies in Fig. 1. The last time-trace represents the total number of neighbors (\(\Sigma\)) within the given cutoff at any given moment in the trajectory. On average, LEU394 has around 4 non-bonded neighbors below the cutoff (see legend of Fig. 1).
Anything that gets shown in any way to the output can be saved for later use as human readable ASCII-files (.dat,.txt), spreadsheets (.ods,.xlsx) or NumPy (.npy) files.
The best way to find out about all of mdciao’s command-line tools is to use a command-line tool shipped with mdciao:
>>> mdc_examples.pyusage: mdc_examples.py [-h] [-x] [--short_options] cltWrapper script to showcase and optionally run examples of thecommand-line-tools that ship with mdciao.Available command line tools are: * mdc_GPCR_overview.py * mdc_CGN_overview.py * mdc_compare.py * mdc_fragments.py * mdc_interface.py * mdc_neighborhoods.py * mdc_pdb.py * mdc_residues.py * mdc_sites.pyYou can type for example: > mdc_interface.py -h to view the command's documentation or > mdc_examples.py interf to show and/or run an example of that command
What these tools do is:
mdc_neighborhoods
Analyse residue neighborhoods using a distance cutoff. Example in Fig. 3.
mdc_interface
Analyse interfaces between any two groups of residues using a distance cutoff. Example here with Fig. 4, Fig. 5, and Fig. 6, among others.
mdc_sites
Analyse a specific set of residue-residue contacts using a distance cutoff. Example in Fig. 8.
mdc_fragments
Break a molecular topology into fragments using different heuristics. Example here.
mdc_GPCR_overview
Map a consensus GPCR nomenclature (e.g. Ballesteros-Weinstein and others) nomenclature on an input topology. Example here.
mdc_CGN_overview
Map a Common G-alpha Numbering (CGN)-type nomenclature on an input topology Example here.
mdc_compare
Compare residue-residue contact frequencies from different files. Example here with Fig. 9.
mdc_pdb
Lookup a four-letter PDB-code in the RCSB PDB and save it locally. Example here.
mdc_residues
Find residues in an input topology using Unix filename pattern matching. Example here.
You can see their documentation by using the -h flag when invoking them from the command line, keep reading the Highlights or the CLI Reference.
paper-ready tables and figures from the command line:
mdc_neighborhoods.pytop.pdbtraj.xtc-rL394--GPCRadrb2_human--CGNgnas2_human-ni-at#ni: not interactive, at: show atom-types
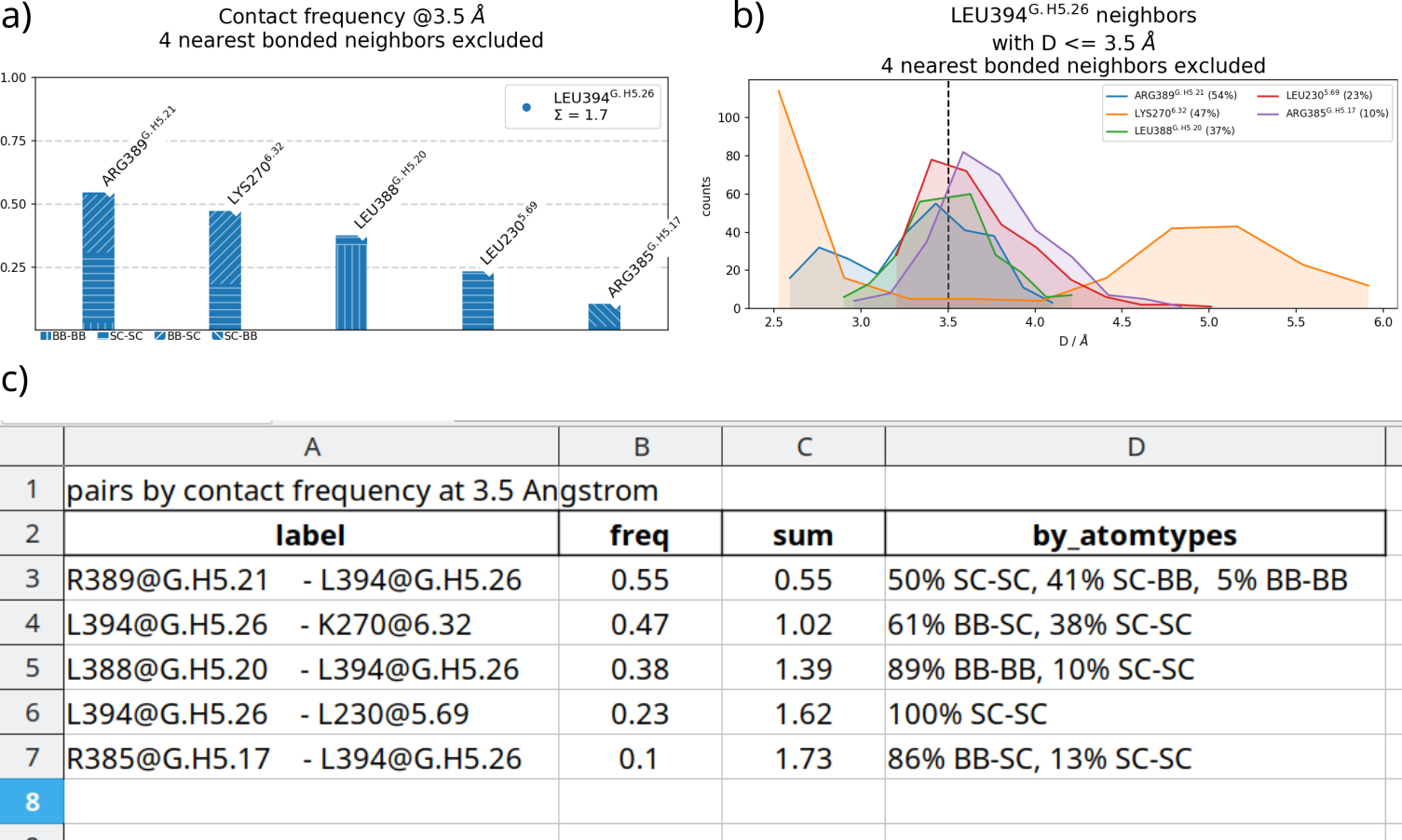
Fig. 3 (click to enlarge) a) contact frequencies for LEU394, as in Fig. 1, but annotated with consensus nomenclature and atom types (--GPCR,--CGN,-at options, see below). b) associated distance distributions, obtained by adding the -d flag to the CLI call. c) Automatically generated table using the -txxlsx option.
automagically map and incorporate consensus nomenclature to the analysis, either from local files or over the network in the GPRC.db and/or KLIFS:
...
No local file ./adrb2_human.xlsx found, checking online in
https://gpcrdb.org/services/residues/extended/adrb2_human ...done!
Please cite the following reference to the GPCRdb:
* Kooistra et al, (2021) GPCRdb in 2021: Integrating GPCR sequence, structure and function
Nucleic Acids Research 49, D335--D343
https://doi.org/10.1093/nar/gkaa1080
For more information, call mdciao.nomenclature.references()
The GPCR-labels align best with fragments: [3] (first-last: GLU30-LEU340).
Mapping the GPCR fragments onto your topology:
TM1 with 32 AAs GLU30@1.29x29 ( 760) - PHE61@1.60x60 (791 ) (TM1)
ICL1 with 4 AAs GLU62@12.48x48 ( 792) - GLN65@12.51x51 (795 ) (ICL1)
TM2 with 32 AAs THR66@2.37x37 ( 796) - LYS97@2.68x67 (827 ) (TM2)
ECL1 with 4 AAs MET98@23.49x49 ( 828) - PHE101@23.52x52 (831 ) (ECL1)
TM3 with 36 AAs GLY102@3.21x21 ( 832) - SER137@3.56x56 (867 ) (TM3)
ICL2 with 8 AAs PRO138@34.50x50 ( 868) - LEU145@34.57x57 (875 ) (ICL2)
TM4 with 27 AAs THR146@4.38x38 ( 876) - HIS172@4.64x64 (902 ) (TM4)
ECL2 with 20 AAs TRP173 ( 903) - THR195 (922 ) (ECL2) resSeq jumps
TM5 with 42 AAs ASN196@5.35x36 ( 923) - GLU237@5.76x76 (964 ) (TM5)
ICL3 with 1 AAs GLY238 ( 965) - ARG239 (966 ) (ICL3)
TM5 with 35 AAs CYS265@6.27x27 ( 967) - GLN299@6.61x61 (1001) (TM6)
ECL2 with 4 AAs ASP300 (1002) - ILE303 (1005) (ECL3)
TM7 with 25 AAs ARG304@7.31x30 (1006) - ARG328@7.55x55 (1030) (TM7)
H8 with 12 AAs SER329@8.47x47 (1031) - LEU340@8.58x58 (1042) (H8)
...
Please cite the following reference to the CGN nomenclature:
* Flock et al, (2015) Universal allosteric mechanism for G$\alpha$ activation by GPCRs
Nature 2015 524:7564 524, 173--179
https://doi.org/10.1038/nature14663
For more information, call mdciao.nomenclature.references()
The CGN-labels align best with fragments: [0] (first-last: LEU4-LEU394).
Mapping the CGN fragments onto your topology:
G.HN with 33 AAs LEU4@G.HN.10 ( 0) - VAL36@G.HN.53 (32 ) (G.HN)
G.hns1 with 3 AAs TYR37@G.hns1.01 ( 33) - ALA39@G.hns1.03 (35 ) (G.hns1)
G.S1 with 7 AAs THR40@G.S1.01 ( 36) - LEU46@G.S1.07 (42 ) (G.S1)
...
G.hgh4 with 21 AAs TYR311@G.hgh4.01 ( 270) - ASP331@G.hgh4.21 (290 ) (G.hgh4)
G.H4 with 16 AAs PRO332@G.H4.01 ( 291) - ARG347@G.H4.17 (306 ) (G.H4)
G.h4s6 with 11 AAs ILE348@G.h4s6.01 ( 307) - TYR358@G.h4s6.20 (317 ) (G.h4s6)
G.S6 with 5 AAs CYS359@G.S6.01 ( 318) - PHE363@G.S6.05 (322 ) (G.S6)
G.s6h5 with 5 AAs THR364@G.s6h5.01 ( 323) - ASP368@G.s6h5.05 (327 ) (G.s6h5)
G.H5 with 26 AAs THR369@G.H5.01 ( 328) - LEU394@G.H5.26 (353 ) (G.H5)
...
easy residue selection, with an extended UNIX-like pattern matching. You can preview residue selections like GLU,P0G,3.50,380-394,G.HN.*, which means:
GLU*: all GLUs, equivalent to GLU
P0G: the B2AR ligand (agonist)
3.50: generic-residue numbering for GPCRs
380-394: range of residues with sequence indices 380 to 394 (both incl.). This is the \(G\alpha_5\)-subunit.
G.HN.* : CGN-nomenclature for the \(G\alpha_N\)-subunit
You can check your selection before running a computation by using mdc_residues.py:
>>> mdc_pdb.py3SN6-o3SN6.groChecking https://files.rcsb.org/download/3SN6.pdb ...doneSaving to 3SN6.gro...donePlease cite the following 3rd party publication: * Crystal structure of the beta2 adrenergic receptor-Gs protein complex Rasmussen, S.G. et al., Nature 2011 https://doi.org/10.1038/nature10361
fragmentation heuristics to easily identify molecules and/or molecular fragments. These heuristics will work on .pdf-files lacking TER and CONNECT records or other file formats, like .gro files, that simply don’t include these records:
In this example, we saved the crystal structure 3SN6 as a .gro-file (mdc_pdb.py3SN6-o3SN6.gro). We are able to recover sensible fragments:
\(G\alpha\)
\(G\beta\)
\(G\gamma\)
bacteriophage T4 lysozyme as N-terminus of the receptor (next)
\(\beta 2\) adrenergic receptor
VHH antibody
ligand.
For clarity, we omitted the fragmentation in our initial example with the option -nf, but all CLI tools do this fragmentation by default. Alternatively, one can use:
mdc_fragments.py3SN6.gro
to get an overview of all available fragmentation heuristics and their results without computing any contacts whatsoever.
use fragment definitions –like the ones above, 0 for the \(G\alpha\)-unit and 3 for the receptor– to compute interfaces in an automated way, i.e. without having to specifying individual residues:
>>> mdc_interface.pytop.pdbtraj.xtc-isel10-isel23--GPCRadrb2_human--CGNgnas2_human-t"3SN6 beta2AR-Galpha interface"-ni...The following 50 contacts capture 45.66 (~91%) of the total frequency 50.28 (over 107 contacts with nonzero frequency at 4.50 Angstrom).As orientation value, the first 50 ctcs already capture 90.0% of 50.28.The 50-th contact has a frequency of 0.52. freq label residues fragments sum1 1.00 R385@G.H5.17 - K232@5.71x71 344 - 959 0 - 3 1.002 1.00 V217@G.S3.01 - F139@34.51x51 183 - 869 0 - 3 2.003 1.00 R385@G.H5.17 - Q229@5.68x68 344 - 956 0 - 3 3.004 1.00 D381@G.H5.13 - Q229@5.68x68 340 - 956 0 - 3 4.005 1.00 E392@G.H5.24 - T274@6.36x36 351 - 976 0 - 3 5.006 1.00 Y358@G.h4s6.20 - S236@5.75x75 317 - 963 0 - 3 6.007 1.00 D381@G.H5.13 - K232@5.71x71 340 - 959 0 - 3 7.00..../interface.overall@4.5_Ang.xlsx./interface.overall@4.5_Ang.dat./interface.overall@4.5_Ang.as_bfactors.pdb./interface.overall@4.5_Ang.pdf./interface.matrix@4.5_Ang.pdf./interface.flare@4.5_Ang.pdf./interface.time_trace@4.5_Ang.pdf
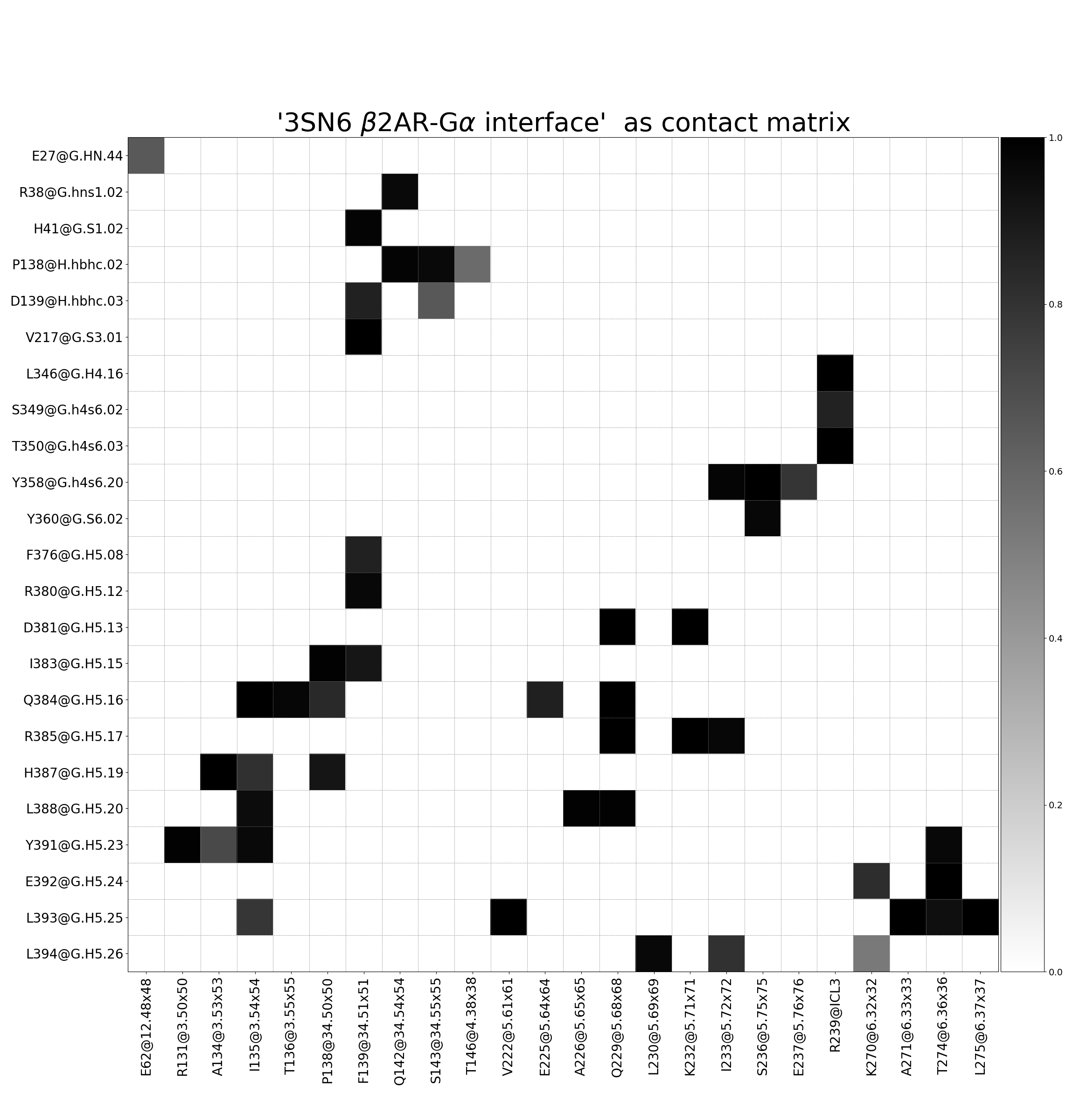
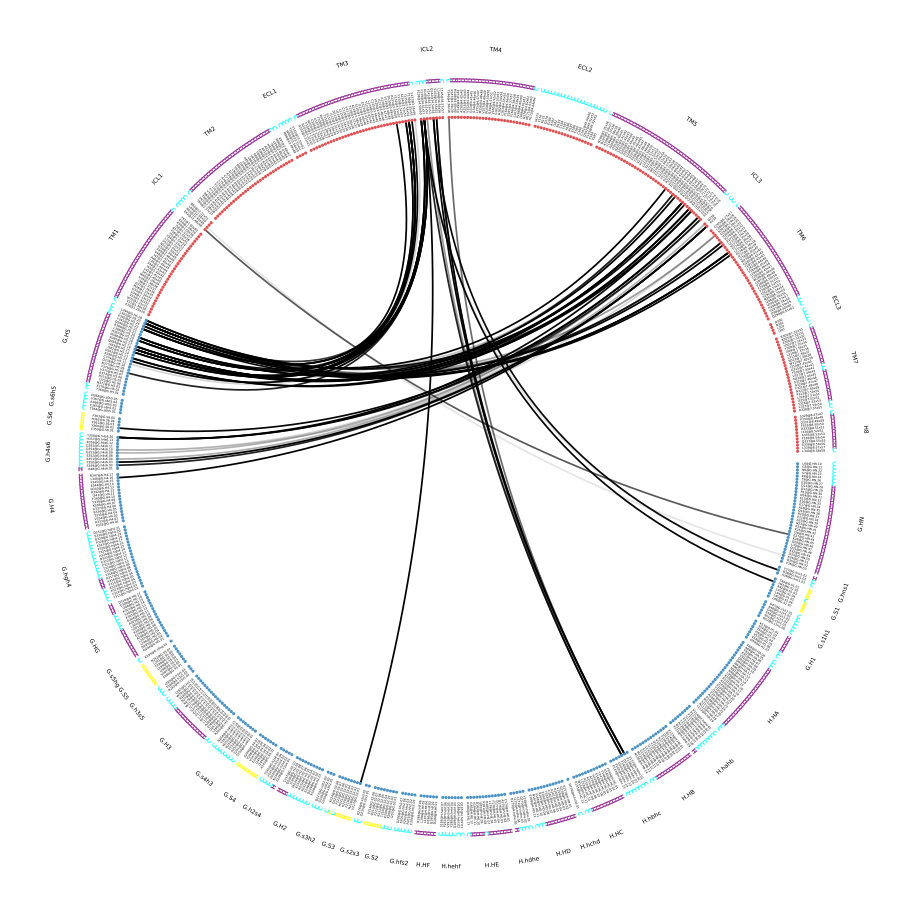
Fig. 4 [interface.matrix@4.5_Ang.pdf](click to enlarge). Interface contact matrix between the β2AR receptor and the α-unit of the G-protein, using a cutoff of 4.5 Å. The labelling incorporates consensus nomenclature to identify positions and domains of both receptor and G-protein. Please note: this is not a symmetric contact-matrix. The y-axis shows residues in the Gα-unit and the x-axis in the receptor.
Since Fig. 4 is bound to incorporate a lot of blank pixels, mdciao will also produce sparse plots and figures that highlight the formed contacts only:
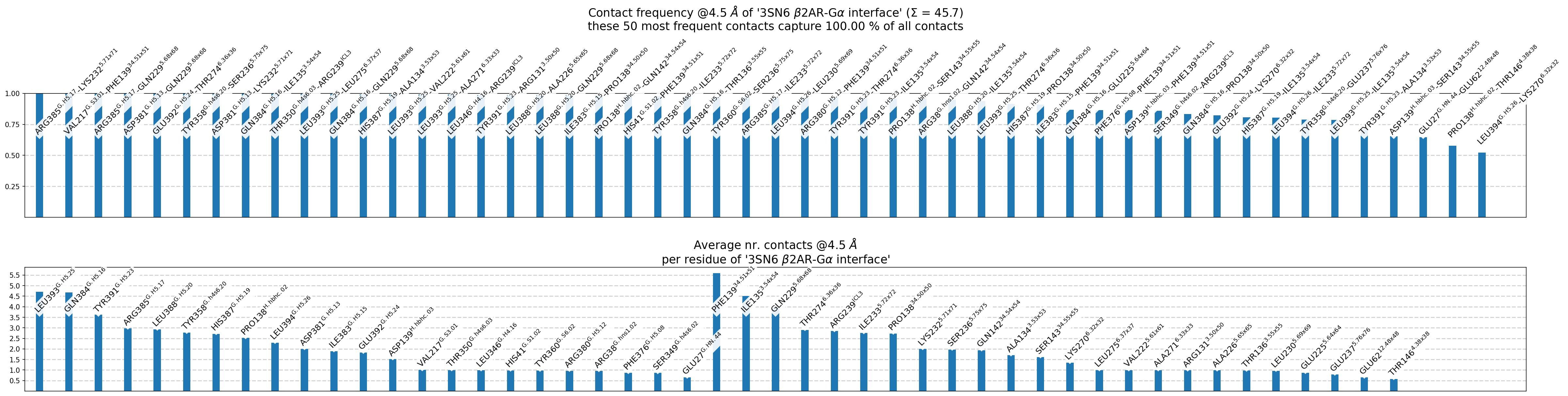
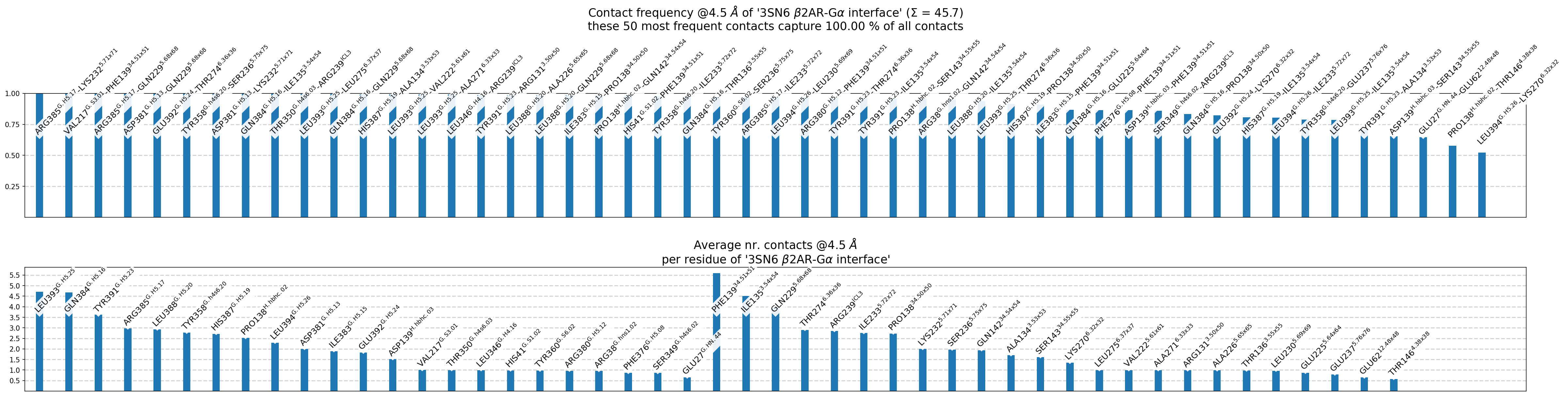
Fig. 5 [interface.overall@4.5_Ang.pdf](click to enlarge) Upper panel: most frequent contacts sorted by frequency, i.e. for each non-empty pixel of Fig. 4, there is a bar shown. Lower panel: per-residue aggregated contact-frequencies, showing each residue’s average participation in the interface (same info will be written to interface.overall@4.5_Ang.xlsx). Also, the number of shown contacts/bars can be controlled either with the –ctc_control and/or –min_freq parameters of mdc_interface.py.
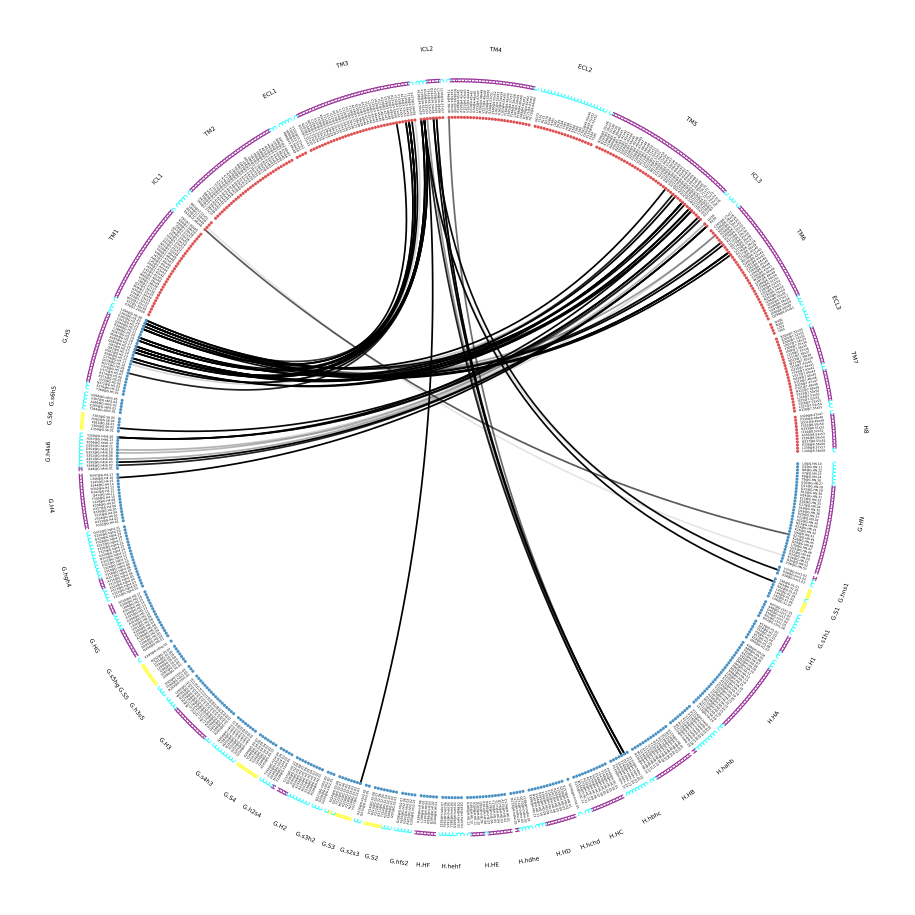
A very convenient way to incorporate the molecular topology into the visualization of contact frequencies are the so-called FlarePlots (cool live-demo here). These show the molecular topology (residues, fragments) on a circle with curves connecting the residues for which a given frequency has been computed. mdciao has its own flareplot implementation in the mdciao.flare module, that can also coarse-grain flareplots to the molecular fragments. The mdc_interface.py example above will generates a flareplot by default:
Fig. 6 [interface.flare@4.5_Ang.pdf](click to enlarge) FlarePlot of the frequencies shown in the figures Fig. 4 and Fig. 5. Residues are shown as dots on a circumference, split into fragments following any available labelling information. The contact frequencies are represented as lines connecting these dots/residues, with the line-opacity proportional to the frequencie’s value. The secondary stucture of each residue is also included as color-coded letters: H(elix), B(eta), C(oil). We can clearly see the \(G\alpha_5\)-subunit in contact with the receptor’s TM3, ICL2, and TM5-ICL3-TM6 regions. Note that this plot is always produced as .pdf to be able to zoom into it as much as needed.
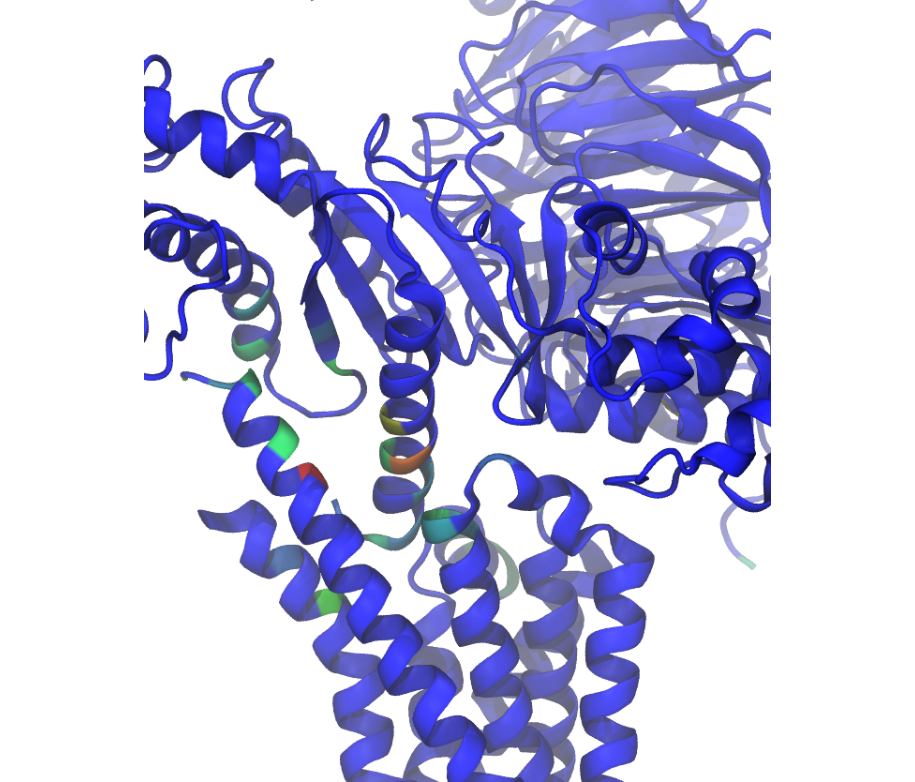
Similar to how the flareplot (Fig. 6) is mapping contact-frequencies (Fig. 5, upper panel) onto the molecular topology, the next figure maps the lower panel Fig. 5 on the molecular geometry. It simply puts the values shown there in the temperature factor of a pdb file, representing the calculated interface as a heatmap, which can be visualized in VMD using the Beta coloring.
Fig. 7 [interface.overall@4.5_Ang.as_bfactors.pdb](click to enlarge) 3D visualization of the interface as heatmap (blue-green-red) using VMD. We clearly see the regions noted in Fig. 6 (TM5-ICL3-TM6 and \(G\alpha_5\)-subunit) in particular the residues of Fig. 5 (lower panel) light up. This heatmap is overlaid on structures representative of the interface, and have been selected using the mdciao.contacts.ContactGroup.repframes method. Please note, for the homepage-banner (red-blue heatmap), the signed_colors argument has been used when calling the mdciao.flare.freqs2flare method of the API. At the moment this is not possible just by using mdc_interface.py, sorry!
You can use this snippet to generate a VMD visualiazation state file, view_mdciao_interface.vmd to view the heatmap:
echo'mol new ./interface.overall@4.5_Ang.as_bfactors.pdbmolmodstyle00NewCartoonmolmodcolor00BetacolorscalemethodBGR' > view_mdciao_interface.vmdvmd-eview_mdciao_interface.vmd
view_mdciao_interface.vmd will work with any *.as_bfactors.pdb file that mdciao generates. For our example, you can also paste this viewpoint into your VMD console and generate a view equivalent to the above picture (results may vary with other files):
A different approach is to look only for a particular set of pre-defined contacts. Simply writing this set into a human readable JSON file will allow mdc_sites.py to compute and present these (and only these) contacts, as in the example file tip.json:
One added bonus is that the same .json files can be used file across different setups as long as the specified residues are present.
The command:
>>> mdc_sites.pytop.pdbtraj.xtc--sitetip.json-at-nf-sa#sa: short AA-names...The following files have been created:./sites.overall@4.5_Ang.pdf...
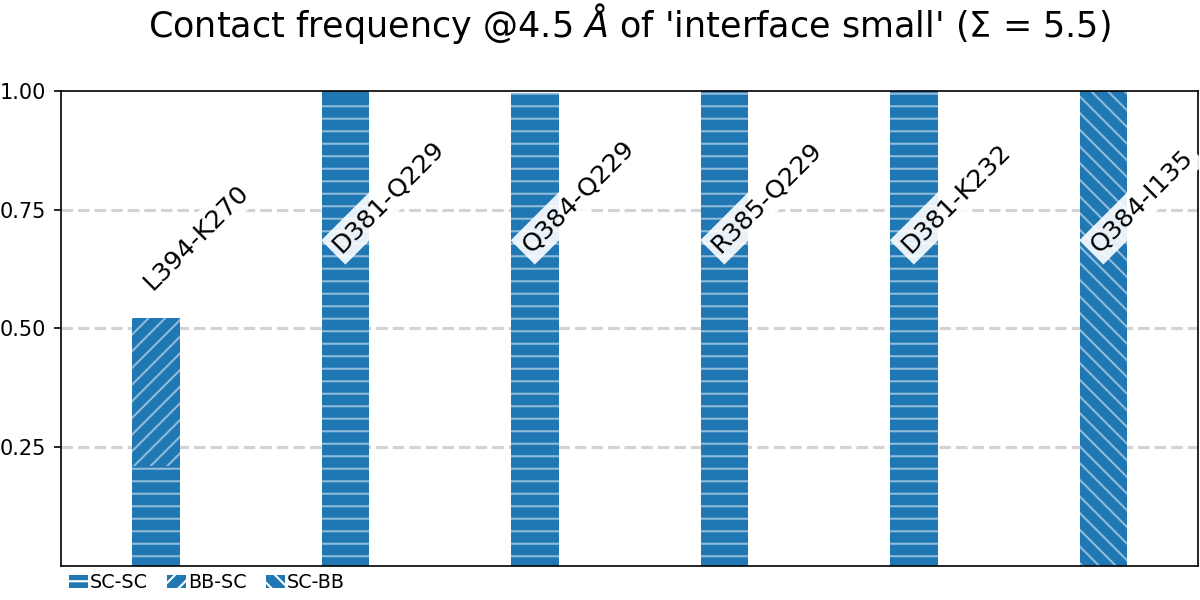
generates the following figure (tables are generated but not shown). The option -at (--atomtypes) generates the patterns (“hatching”) of the bars. They indicate what atom types (sidechain or backbone) are responsible for the contact:
Fig. 8 [sites.overall@4.5_Ang.pdf](click to enlarge) Contact frequencies of the residue pairs specified in the file tip.json, shown with the contact type indicated by the stripes on the bars. Use e.g. the 3D-visualisation to check how “L394-K270” switches between SC-SC and SC-BB.
compare contact frequencies coming from different calculations, to detect and show contact changes across different systems. For example, to look for the effect of different ligands, mutations, pH-values etc. In this case, we compare the neighborhood of R131 (3.50 on the receptor) between our MD simulations and the crystal structure straight from the PDB. First, we grab the file on the fly with mdc_pdb.py:
>>> mdc_pdb.py3SN6Checking https://files.rcsb.org/download/3SN6.pdb ...doneSaving to 3SN6.pdb...donePlease cite the following 3rd party publication: * Crystal structure of the beta2 adrenergic receptor-Gs protein complex Rasmussen, S.G. et al., Nature 2011 https://doi.org/10.1038/nature10361
Now we use mdc_neighborhoods.py on it:
>>> mdc_neighborhoods.py3SN6.pdb3SN6.pdb-rR131-o3SN6-nf-o3SN6.X...The following 5 contacts capture 5.00 (~100%) of the total frequency 5.00 (over 5 contacts with nonzero frequency at 4.50 Angstrom).As orientation value, the first 5 ctcs already capture 90.0% of 5.00.The 5-th contact has a frequency of 1.00. freq label residues fragments sum1 1.0 R131 - Y326 1007 - 1174 0 - 0 1.02 1.0 R131 - Y391 1007 - 345 0 - 0 2.03 1.0 R131 - V222 1007 - 1095 0 - 0 3.04 1.0 R131 - Y219 1007 - 1092 0 - 0 4.05 1.0 R131 - I278 1007 - 1126 0 - 0 5.0The following files have been created:./3SN6.X.ARG131@4.5_Ang.dat
Now we use mdc_neighborhoods.py on our data:
>>> mdc_neighborhoods.pytop.pdbtraj.xtc-rR131-nf-o3SN6.MD...The following 6 contacts capture 3.15 (~99%) of the total frequency 3.17 (over 7 contacts with nonzero frequency at 4.50 Angstrom).As orientation value, the first 4 ctcs already capture 90.0% of 3.17.The 4-th contact has a frequency of 0.39. freq label residues fragments sum1 0.99 R131 - Y391 861 - 350 0 - 0 0.992 0.94 R131 - Y326 861 - 1028 0 - 0 1.923 0.76 R131 - Y219 861 - 946 0 - 0 2.694 0.39 R131 - I278 861 - 980 0 - 0 3.075 0.05 R131 - I325 861 - 1027 0 - 0 3.126 0.02 R131 - I72 861 - 802 0 - 0 3.15...The following files have been created:..../3SN6.MD.ARG131@4.5_Ang.dat
Please note that we have omitted most of the terminal output, and that we have used the option -o to label output-files differently: 3SN6.X and 3SN6.MD. Now we compare both these outputs:
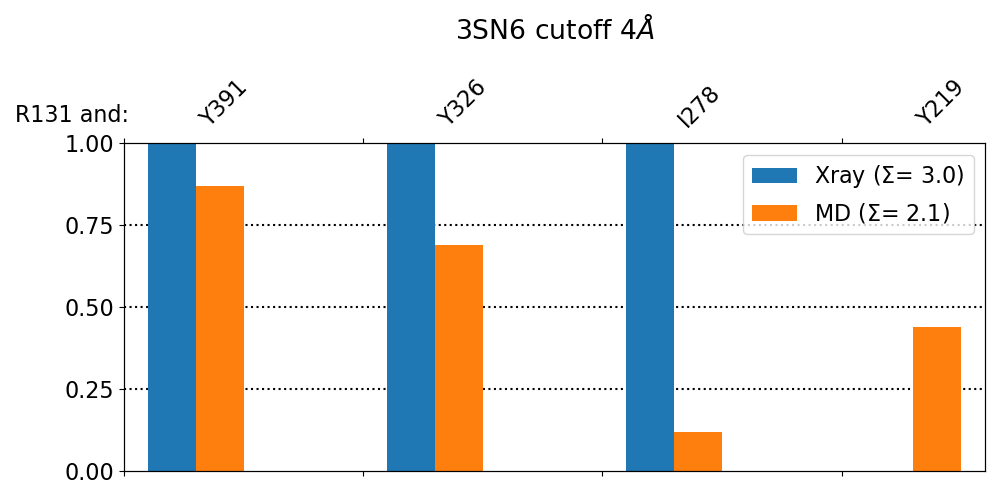
>>> mdc_compare.py3SN6.X.ARG131@4.5_Ang.dat3SN6.MD.ARG131@4.5_Ang.dat-kXray,MD-t"3SN6 cutoff 4.5AA"-aR131These interactions are not shared:I325, I72, V222Their cumulative ctc freq is 1.07.Created filesfreq_comparison.pdffreq_comparison.xlsx
Fig. 9 [freq_comparison.pdf]Neighborhood comparison for R131 between our MD simulations and the original 3SN6 crystal structure. We can see how the neighborhood relaxes and changes. Some close residues, in particular I278, move further than 4.5 Å away from R131. You can see these residues highlighted in the 3D visualization. We have used a custom title and custom keys for clarity of the figure (options -t and -k). Also, since all contact labels share the ‘R131’ label, we can remove it with the -a (anchor residue).
The API is what you get to your Python scope upon importing mdciao, .e.g.:
importmdciao
The API has a dedicated submodule to replicate the Command-Line-Interface, mdciao.cli and a
lot of other methods that expose useful functions to create your own workflows. The the Jupyter Notebook Tutorial
has some highlights and easy walkthroughs, while the Jupyter Notebook Gallery for some more fancy examples.
Check the API Reference page for the full API documentation.
Note
Whereas the command-line-tools tend to be more stable, the API might experience some minor changes until the first major release.