Methods
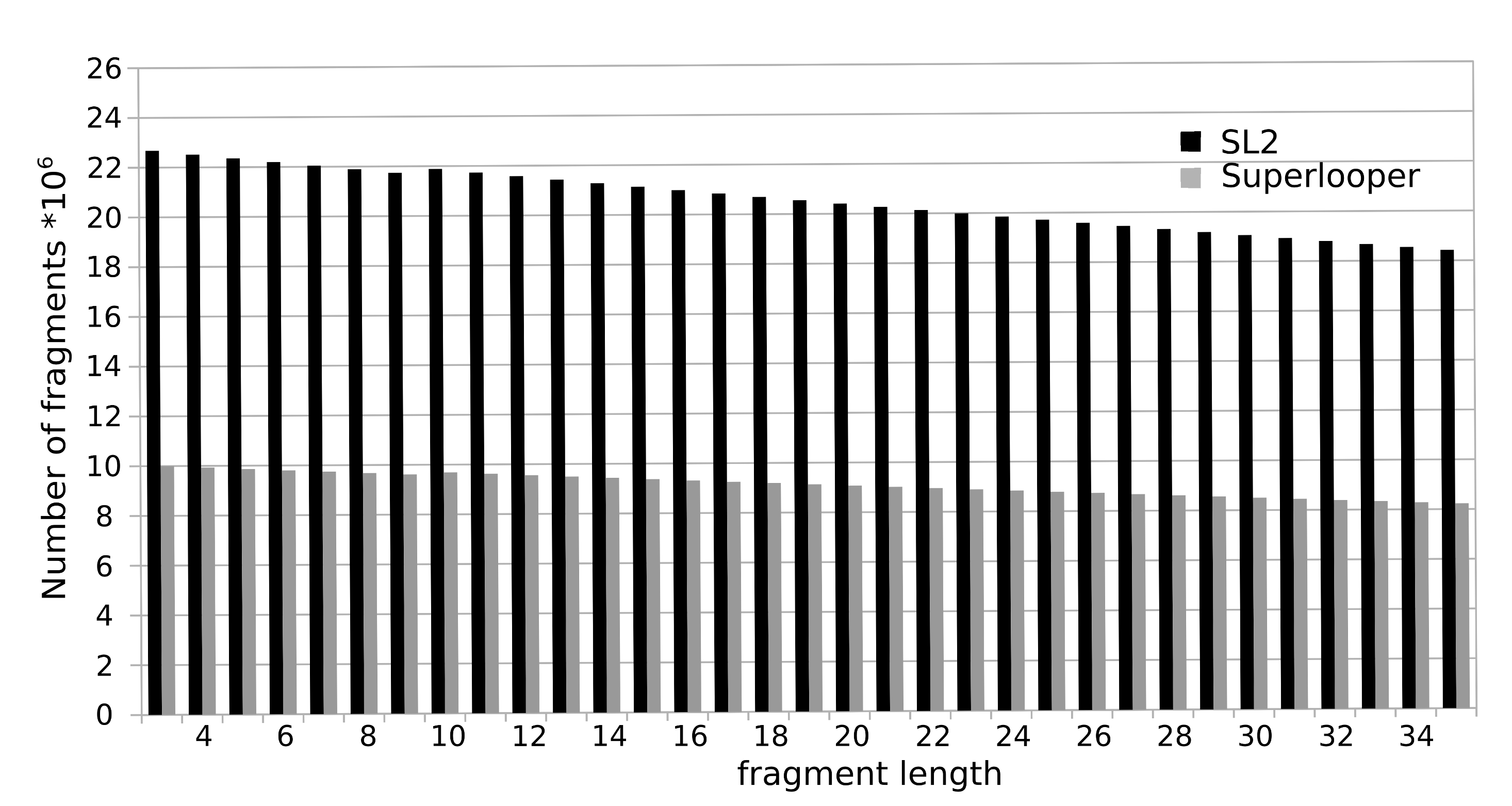
Database
To construct the database all overlapping fragments with a length of 3 to 35 were extracted from a collection of more than 100,000 protein structures currently deposited in the RSCB PDB. For each fragment the following information is stored in the database: sequence, PDB identifier, location in the protein and a geometrical fingerprint (see below). The total number of fragments sum up to over 700 million. The number of available fragments is decreasing with increasing number of residues. This is caused by the fact that a smaller number of long fragments can be extracted from a structure, e.g. for the fragment length of 3 amino acids 23 million fragments are available, as for a fragment length of 35 this number declines to approximately 18 million.
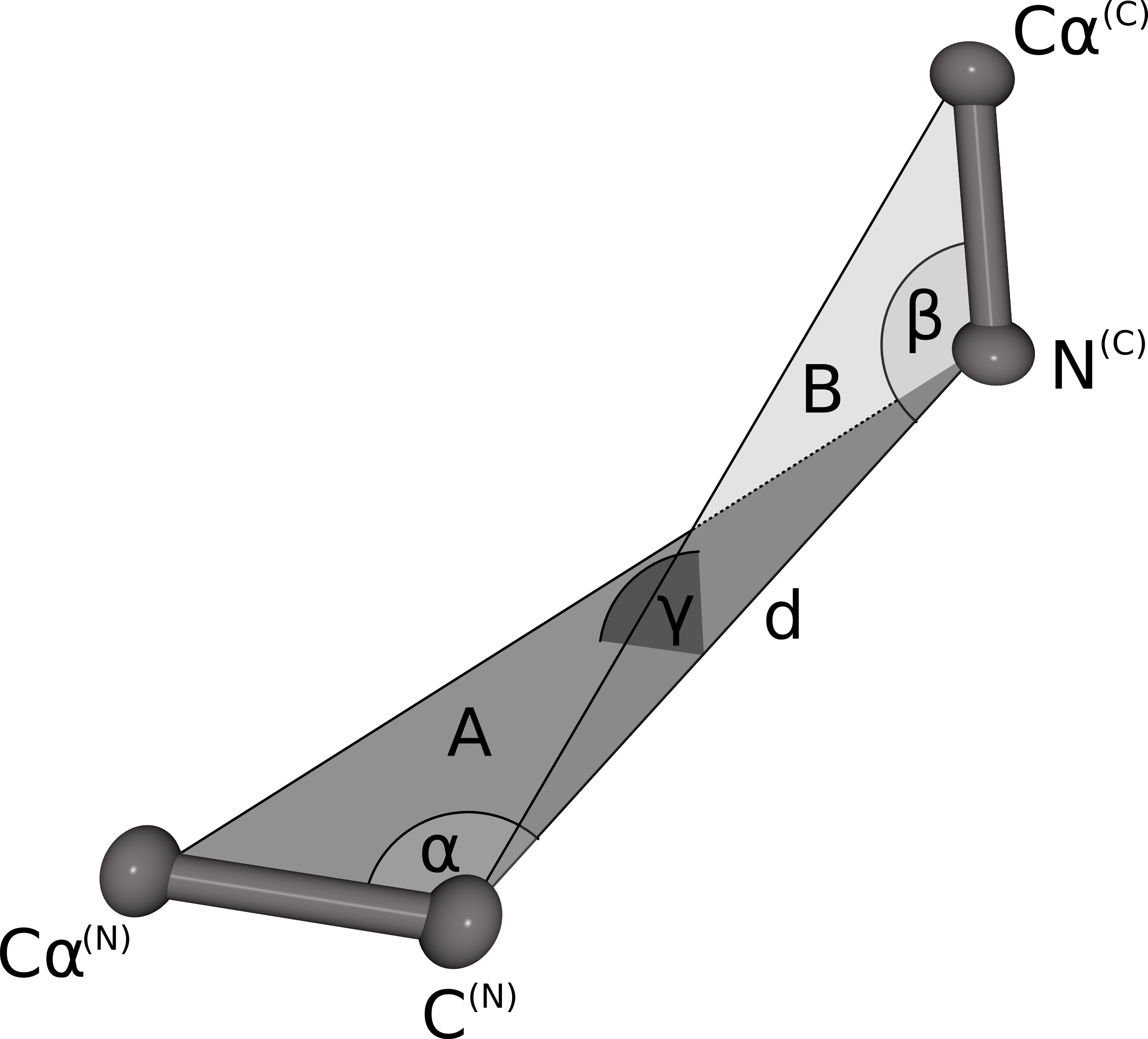
Geometric fingerprint
The geometrical fingerprint matching is used to evaluate the sterical fit of the stem atoms of the N- and C-termini of each database fragment to the C- and N-terminal stem atoms of a gap in the protein structure. Both geometrical fingerprints are composed by the distance between the N- and C-terminal stem atoms, and three angles defining the relative orientation of the stem atoms. The geometrical fingerprint is characterized by the distance d between the N-terminal C atom and the C-terminal N atom, and the following three angles: α defined by the line between Cα(N) and C(N) and d, β is spanned by the line between N(C) and Cα(C) and d, γ defines the angle between the two planes A (defined by Cα(N), C(N)) and N(C)) and B (Cα(C), C(N) and N(C)). This fingerprint is a slight alteration of the fingerprint used in our previous publication. Instead of combining two distances and two angles we now use one distance and three angles. Analysis of the previous fingerprint revealed that the score was slightly biased towards the residue where the angle was measured. By using angles on both stem residues, the resulting score does not favor the fit of the candidate fragment to one stem residue over the other any more.
Score
The 500 best loop candidates are ranked according to a score that is composed by a measure for the sequence identity and the fit of the stem atoms of loop and gap.score = M - 0.1 * RMSD_stem2
The sequence score M is calculated using an environment-specific amino acid substitution matrix for accessible residues. The stem score RMSD is the root mean square deviation of the stem atoms Cα and C of the N-terminal stem residue and N and Cα of the C-terminal stem residues.
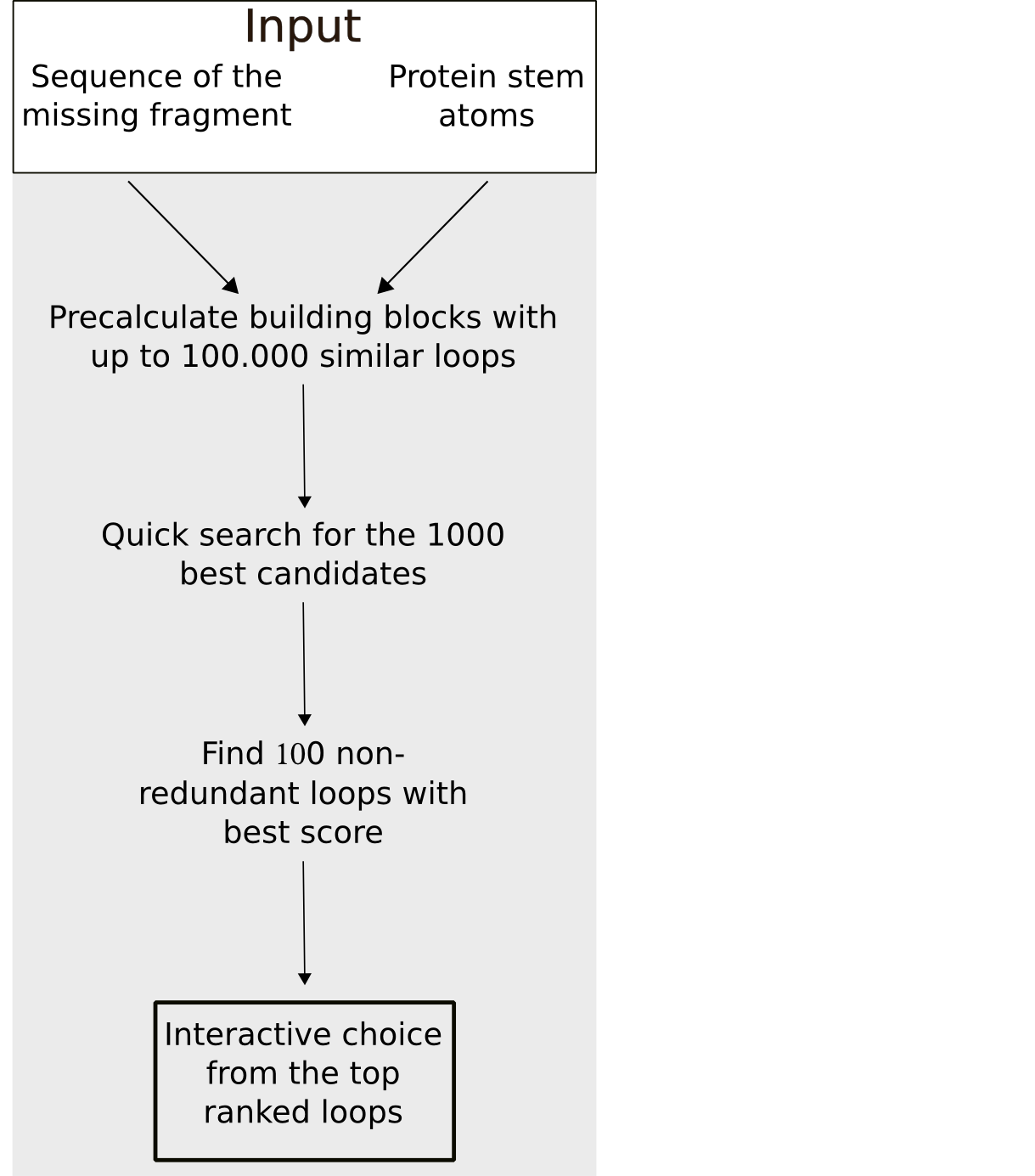
Fragment Search
To minimize the calculation time of the fragment search a stepwise approach is used. In a first step, all fragments with the defined sequence length and whose stem atoms match the stem atoms of the gap with at least 0.75 Å RMSD are selected. In the second step, the 500 top candidates are chosen based on a quick estimation of the steric fit of the fragment to the rest of the protein e.g. excluding clashes. Subsequently, these 500 fragment are ranked by a score calculated from the sequence similarity and matching of the geometrical fingerprints of the (template) fragment and the target segment. To maximize the conformational space, fragments with an identical sequence or a backbone RMSD smaller than 1 Å are removed from the results list.
Membrane planes
For the calculation of the membrane planes we employ the web-service TMDET. As result of the calculation, TMDET [3] provides a list of residues that tangent the membrane. From coordinates of Cα atoms of these residues we then calculate a best-fit plane which is displayed in the NGL viewer.
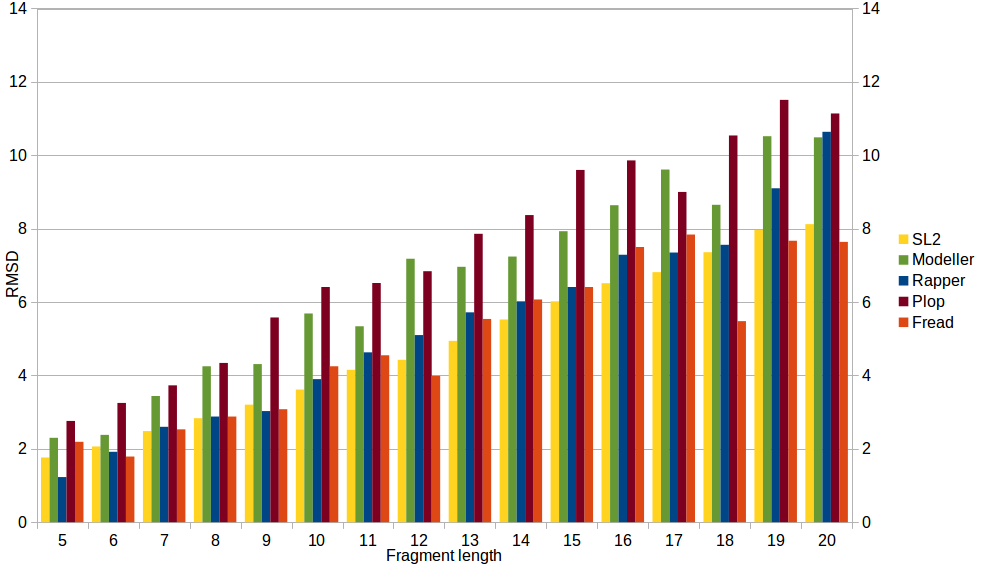
Tool comparison
To compare performance of SL2 with other modeling tools, the RMSD values of RAPPER, PLOP, MODELLER and FREAD were generated applying the reference dataset used in Choi and Deane, Proteins 2009 and the RMSD values of SL2 (method & LIP database updated in 2016) were generated applying the reference dataset used in Michalsky, Goesde, Preissner, Protein Eng. 2003. SL2 performs significantly better (independent of length) than modeller and Plopn and comparable to Rapper and Fread.