Frequently Asked Questiones
Import data
-
How do I upload structures?
At the top of the main web page, click the upload structure and mutations menu button. This displays a form which offers three ways to upload a structure:
- from the Protein Databank(PDB)
- from the AlphaFold-EBI website
- or from your local machine
To use an AlphaFold model, enter either:
- the UniProt ID (e.g. Q969P0)
- the full AlphaFold ID (e.g. AF-Q969P0-F1)
- or the full filename (e.g. AF-Q969P0-F1-model_v4.pdb).
(UniProt and AlphaFold IDs are automatically extended to download the corresponding AlphaFold model.)
-
Can I upload data from sequencing experiments?
Yes, you can upload files in VCF format (https://docs.gdc.cancer.gov/Data/File_Formats/VCF_Format/).
However, only human SNP data can be evaluated currently. Further restrictions are listed on screen under upload vcf.
-
How are VCF uploads processed?
VCF files are forwarded to the ENSEMBL Variant Effect Predictor (GRCh38 version 105). The returned missense variants are filtered to retain only ENST transcript IDs that directly cross-reference to curated UniProtKB/Swiss-Pro UniProt IDs. To ease AlphaFold integration, only records that refer to canonical UniProt sequences with length ≤ 2700 are processed further. From the UniProt ID, the AlphaFold model is automatically selected. The model and its computed missense variants are together submitted to MutationExplorer.
-
How do I know if my structure has already been minimized and is in the database?
During mutation selection after structure upload or specification, a message is displayed whether the structure has already been minimized:
- PDB entry
PDB IDhas already been minimized in our database. No additional minimization will be performed. - PDB entry
PDB IDwas not previously minimized in our database. Minimization will be performed.
It is not possible to overwrite the minimization in the database.
- PDB entry
-
Can a structure that is within the database be overwritten?
No, structures within the database can not be overwritten.
Minimization
-
How are the structures minimized?
The minimizations we offer as 'long' and 'short' are both only side-chain optimizations. However, we recommend you to perform the following minimization. It is optimizing also the backbone in a limited way (due to '-relax:constrain_relax_to_start_coords'). A free backbone minimization might be leading to problems for e.g. membrane proteins.
PATH_TO_ROSETTA/relax.static.linuxgccrelease -relax:fast true -relax:cartesian true -score:weights ref2015_cart -use_input_sc -optimization::default_max_cycles 200 -linmem_ig 10 -relax:constrain_relax_to_start_coords -ex1 -ex2 -nstruct 20 -in:file:s INPUT.pdb -out:pdb -out:prefix OUTDIR/This commandline will create 20 models, from which you should select the one with the lowest energy. Each PDB contains a line starting with 'pose'. In this line you want the last value, which is the total energy.
-
How are the structures in the database minimized?The structures in the data base are minimised with the option "long".
Mutations
-
How can I define mutations?
Once you submit a structure, you are forwarded to the mutation definition page. Three options are offered for defining mutations (click on the field to open the actual sub-form):
- Define any number of mutations manually using the simple syntax explained via the info button text (also in an FAQ below). There is also an info button available PDB chains which lists all possible residue IDs for the existing chains.
- Upload a sequence alignment (ClustalW format)
- Provide a target sequence and select the chain in the PDB to which the target sequence belongs. You can extend the form to have three input fields for each of theseupload methods:
- Fasta file upload: The fasta file should only contain one sequence and must beassigned to one of the chains. For each chain, only one sequence can be assigned.
- Paste sequence: The sequences must have the same length as the protein chain it is assigned to.
- UniProt ID: The sequence will be retrieved directly from the UniProt website.
There is no limit to the number of simultaneous mutations which can be explored.
-
What is the syntax for manually defining mutations?
Each amino acid mutation must be input in the simple yet strict format
X:nnnAwhere:Xis the chain as given in the PDB file, followed by the:separatornnnis the residue numberAis the one letter code of the new (standard) amino acid to which you are mutating the residue.
Separate multiple mutations with commas. Do not enter spaces.
Example: A:12G,B:123W defines two simultaneous mutations:
- Chain A residue 12 will be mutated to glycine.
- Chain B residue 123 will be mutated to tryptophan.
-
How do I know which positions are possible for manually defining mutations?
In the Manual mutation definition tab you can find a button available PDB chains. A window will open in which you will find all possible chains and their ranges on which mutations are possible.
-
How do I use an alignment to define mutations?
There are two reasons for using alignments. First, an alignment can more easily define potentially many more mutations, more efficiently, than a manual input. Second, the differences in structure and energy provide deep insights into the alignment itself. Only aligned positions can be used to define mutations. Gaps cannot be translated into mutations. Thus, one sequence in the alignment must exactly match one chain in your PDB.
It’s easy to upload an alignment. Click upload structure and mutations, upload a structure and then select to upload an alignment file in ClustalW format.
-
Is it possible to mutate peptides?
Yes, it is. MutationExplorer has no restrictions concerning the size of uploaded proteins. However, when interpreting calculated energies, be mindful of limitations (see FAQ ”Limitations”). Energies in flexible regions should be interpreted with particular care, andshorter peptides are more likely to be flexible. Moreover, every protein conformation is just one snapshot. The smaller the peptide, the more likely a given conformation might not well represent the effect of mutation vis-à-vis an alternate conformations.
-
Is it possible to mutate transmembrane proteins?
In general, yes. However, the Rosetta energy function that we use is optimized for soluble proteins and does not consider the lipid bilayer.
-
What about nonstandard amino acids, lipids, ions?
Currently, nonstandard amino acids are not supported as mutation targets. Further limitations apply to disulfid bonds. Nevertheless, nonstandard amino acids occur frequently in the PDB (along with all manner of non-amino acid molecular species). MutationExplorer will consider hetero atoms such as water, ions or ligands when performing mutations. However, we recommend removal of ions or water molecules so that variant residues can fit more easily into the structure. In general, small molecules such as lipids, drugs, ions, or water, are not included in MutationExplorer calculations.
-
How do you mutate a mutant (perform multiple rounds of mutations)?
Once you have performed your first mutations, the results will be displayed along with a window where you can enter further mutations to the variant structure.
- In the Select region, select the variant you want to mutate. The displayed protein is mutated in the next step.
-
In the Mutate section, define any number of mutations.
As always, you will use the
X:nnnAmutation syntax explained in the FAQ above.
-
Is it possible to mutate the same residue multiple times?
Yes, it is possible to mutate one and the same amino acid several times in succession. However, it still leads to errors from time to time. If you encounter an error, you can send us your job id with your error by e-mail.
-
Can I run without defining a mutation in the input?
No, this is currently not possible but might be added in the future. If you want to simulate such a behavior, add a silent mutation.
-
Why do I see an energy difference when making silent mutations?
Every time we perform the mutations and calculate the energy, the structure will be relaxed. this can lead to changes in rotamers, which can affect the total energy of the structure. Those should normally only be minor.
Explorer
-
According to which values can the structure be colored?
From the Color by drop-down in the Select area (top left in the result page), you can choose the following colorings:
- Absolute rosetta energy
- Energy difference to parent
- Absolute hydrophobicity
- Hydrophobicity difference to parent
- Absolute interface score energy
- Interface score energy difference to parent
- Conservation
For energy values, the color scale ranges from blue (negative values) over white (zero) to red (positive values). Restated, red represents increased energy from mutation(s), blue represents decreased energy, and white conveys no energetic change.
For hydrophobicity coloring, the color scale ranges from blue (negative) over white (zero) to yellow (positive). Yellow indicates a hydrophobic residue, blue hydrophilic.
The color scales are shown in detail below in the tab "What are the values of the color scale?".
-
What is your naming convention for structures?
mut_0.pdb is the original protein without mutation. Subsequent naming reflects the tree-like progress of explorations which grow from the mut_0 root. You can select any listed file to mutate it.
- mut_0_1 is the first mutation of the original protein.
- mut_0_1_1 would be a further mutation of the first mutant (mut_0_1).
- mut_0_2 is the second independent mutation of the original protein mut_0.
-
Why is not possible to display energy differences for mut_0.pdb?
The energy difference can only be displayed for mutants. mut_0.pdb is the original protein and has no parent protein to which it could be compared to.
-
What do I do if my structure is no longer visible?If your structure was visible before, try reloading the page, toggling which structure should be visible, or changing the coloring. If our structure was never visible, some files may be missing. Please write us your Job ID so we can investigate further.
Viewer
-
How can I take a screenshot of the viewer?
This is possible by clicking on the screenshot button (see image below). The settings for the resolution, the representation of the axes and the background can also be set in the screenshot menu. Click on the download button to export the screenshot from MutationExplorer.
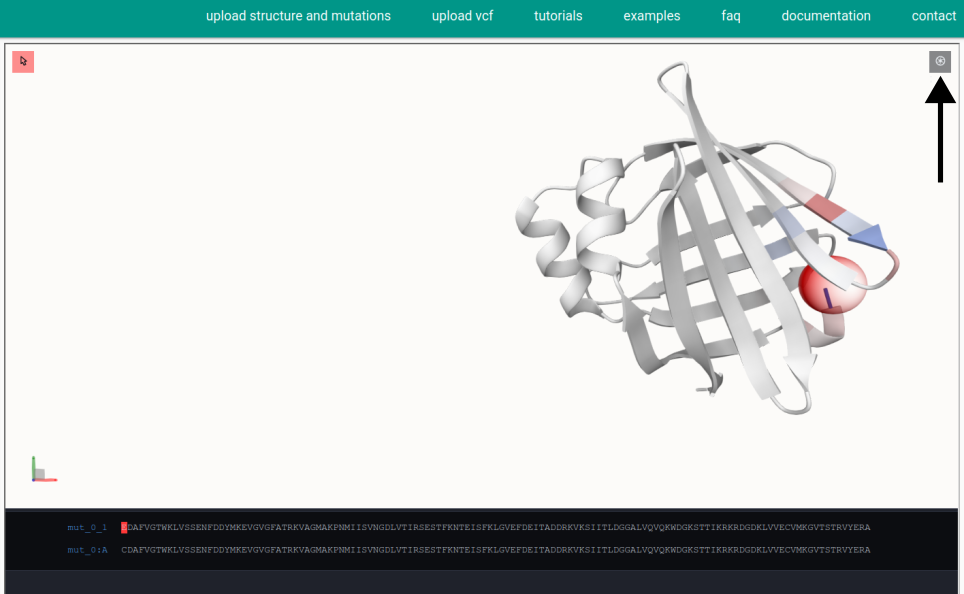
Energy color scale -
How can I change the representation of the structures?
Currently, we only offer a cartoon representation for the structures. We want to include more representations in the future.
-
I don’t see an alignment - why?
If you do not see an alignment at the bottom, this may be due to two possible sources of error. At least one sequence in the clustal file must be identical or part of the sequence of a chain in the uploaded structure. The clustal sequences are checked against the sequences in the structure so that they can be displayed. The second source of error could be that our internal matching did not work properly. If you do not see an alignment, please send us an e-mail with your job ID so that we can solve the problem.
-
What do I do if the wrong structure is highlighted when hovering over the sequence in the Alignment View tab?
This is an error that should not occur. However, if it does, there seems to be a problem with the internal matching of the structure to the alignment. Please send us your Job ID so that we can find the error.
-
What do I do if the mutation sphere is located on a wrong amino acid?
This is a bug related to the question directly above. Something went wrong in the internal matching, resulting in the wrong amino acid being highlighted as the mutation site. Please send us your Job ID so that we can find a solution.
-
What are the values of the color scales?
Energy and hydrophobicity
The default color scale ranges from -2 over 0 to +2. You can change the intervals for the color scale in the Change Visualization tab.
Energy color scale 
Hydrophobicity color scale Interface score
The interface score has 5 different bins:
Conservation
- red - gap
- dark blue - a mutation between amino acid groups
- light blue - a mutation withing an amino acid group
- white - no mutation/identical

-
Can I change the interval of the color scale?
You can change the intervals for the color scale (energy and hydrophobicity) in the Change visualization tab.
You cannot change the color scale for the conservation and interface score.
-
How can I open the RaSP prediction window?
You can open the RaSP window for a specific amino acid with Ctrl + right click or by switching on the RaSP selection mode. This can be done by activating the button in the top left hand corner of the viewer. When the selection mode is on, you can just select an amino acid with a left click and the window will open.
- red - off
- green - on
-
Why do I see some weird residues in the structure?
Normally, those are hetero atoms that have not been removed. If you observe some other strange visualizations, please reach out to us.
-
Is it possible to see the residue that was present before the mutation took place?
Yes, you can make the original residue visible as a highlight.
- Select Change Visualization at the bottom of the viewer.
- Select the button saying Hiding highlight of original residue to enable the highlighting.
- Hiding highlight of original residue means the highlight is currently disabled.
- After selecting the button, it will now say Visible highlight of original residue.
- Do not click on the Apply button above! It is connected to the color scale!
- Now select a residue where you want to see the highlight.
- When a parent is available, its residue will now be shown as a highlight.
- Otherwise you will only see the ball and stick representation of the seleced part.
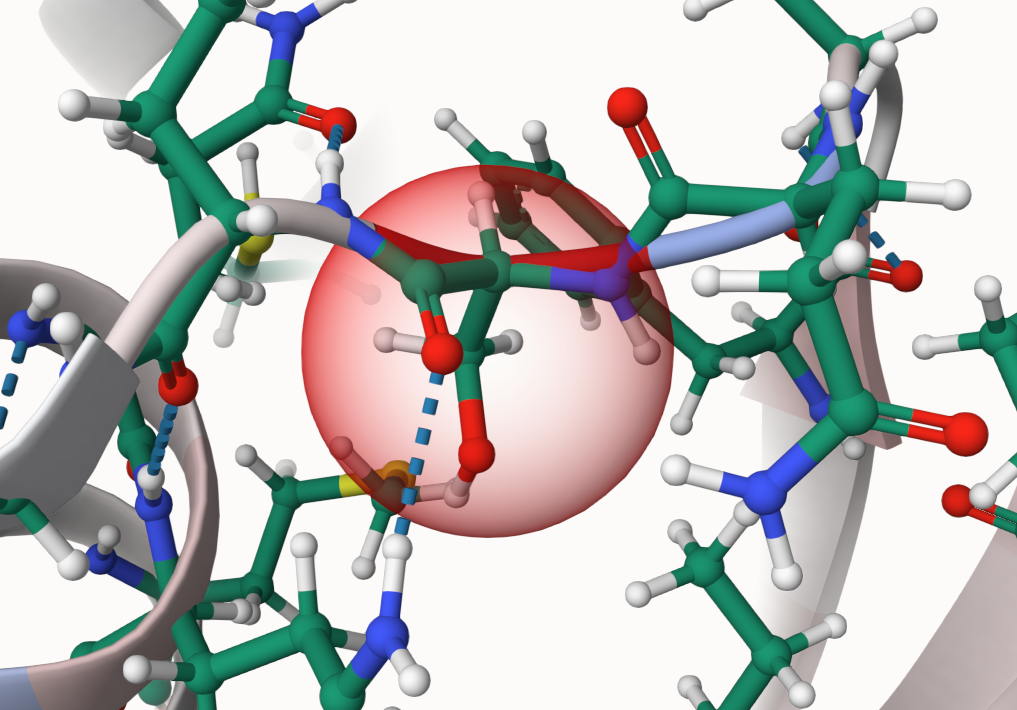
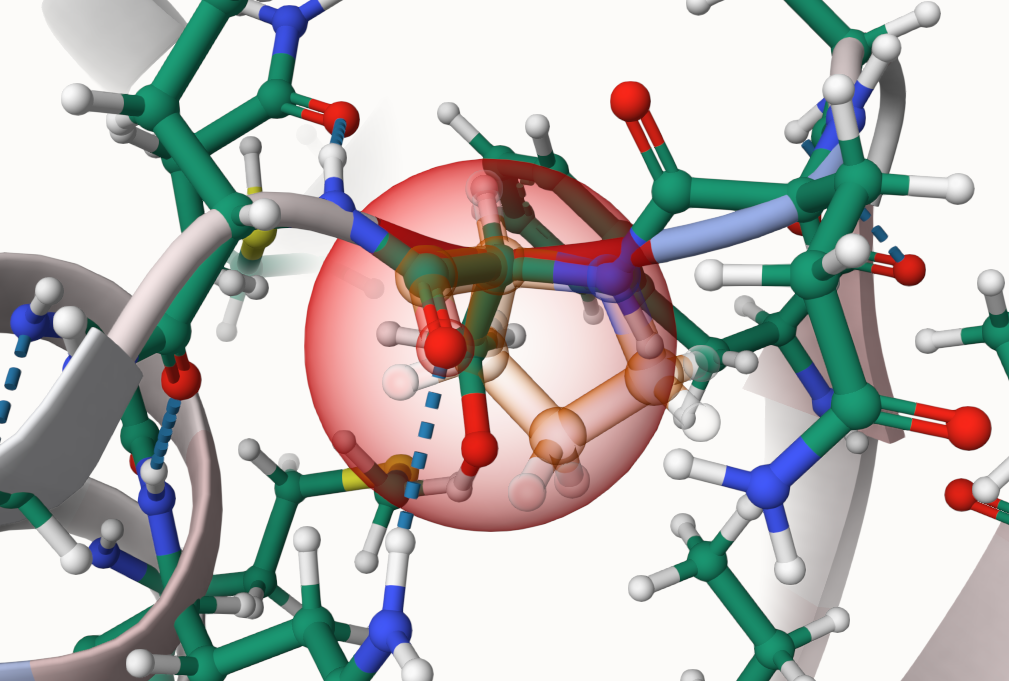
-
The detailed representation does not show anymore when I select a residue after I changed something in the Change Visualization tab.
If this happens, please reload the page. It should then work normally.
Other
-
How does the interface score work?Binding score
InterfaceAnalyzer calculates the binding energy by scoring the input structure twice: first, as a complex, and second, after moving the two sides of the interface away from each other, exposing the interface. The idea is that energy score of the residues that are interacting with residues on the other side of the interface is going to change after those residues are exposed. The Rosetta energy scores of the unbound state are then subtracted from the scores of the bound one. Both the bound (input structure provided by user) and unbound structures have their side chains optimized prior to scoring. Since side chain optimization is stochastic, the mover is run 3 times, and the median score is taken for every residue.
Interface selection
InterfaceAnalyzerMover which we use for the binding score calculation takes as an input a list of all chains in the structure, with interface sides separated by_ . For example, for the protein complex with chains A, B, and C, you can select interfacesA_BC ,B_AC , andC_AB . Which side is which does not affect the score, soA_BC is the same asBC_A . You can specify the interface yourself choosing which chains goes to which side, or you can choose an All vs all mode, which will result in great increase of the runtime. In this mode, the InrefaceAnalyzer is run separately for every chain, consecutively specifying one of the chains as the left side of the interface, with all other chains being assigned as the right side. Thus, the user can see binding dG values for all of the interface residues for the entire structure.
Calculation time
1. If you provide the interface, the addition to the overall running time should be under 5 minutes, depending on the number of chains and the size of the structure. 2. If you choose the all vs all option to see all possible interfaces in the structure, the running time heavily depends on the number of chains, because IntefaceAnalyzer will run for every chain separately. Our estimate runtime for a complex with averagely sized (~300 residue) chains is about N_chains * 5, plus the overall running time. -
Does MutationExplorer handle multichain proteins?
Yes. MutationExplorer always performs calculations in global protein complex context. You must take care to match alignments to specific chains, and specify the correct chain ID when inputting manual mutations. (Presently, VCF inputs are only displayed automatically on single-chain AlphaFold models).
-
How are the energies calculated?
Rosetta performs the mutation and calculates the energy of a structure as a the sum of energies computed for each residue. In the difference coloring, we see the mutant (mutated result structure) minus the WT (parent structure) energy for each residue. Be aware that those energies are indicative and should be revisited with care. For qualitative values that reflect experimental data, other, more time-intensive protocols should be used, e.g. https://pubmed.ncbi.nlm.nih.gov/36173174/
-
Why do some of my mutations exhibit high energies?
MutationExplorer is a platform which must calculate and present energy differences to users in near real-time. Minimizing the protein backbone is not possible given the tight run-time constraints of the application. Thus, mutations can cause in-silico clashes which cannot be resolved without more exhaustive sampling and relaxation techniques. In particular, mutations of residues from or to proline or glycine tend to greatly challenge Rosetta’s minimizer.
-
Why do some distant residues have a change in energy?
When a mutation is introduced, side-chain optimization is not limited to immediately adjacent residues. As the minimizer descends the energy landscape of the entire protein, distant side-chains can be re-oriented by the algorithm.
-
Which parameters were used in the examples?
Each example includes all settings and files needed to re-run and replicate the example outputs. The three examples in the first row demonstrate upload structure and mutations. The fourth example in the second row demonstrates upload vcf.
-
Where can I find more information about the tools used on the server, i.e. MDsrv/mol*/Rosetta?
MDsrv:
https://proteininformatics.informatik.uni-leipzig.de/mdsrv
mol* viewer:
https://molstar.org/
Rosetta:
https://www.rosettacommons.org/
Varient Effect Predictor:
https://www.ensembl.org/info/docs/tools/vep/
General
-
How long will a run take?
Actual times depend on the size of the system and may vary significantly.
- initial basic minimization: 1-3 min
- per round of mutations: \~2 min
- VCF processing (plus minimization): 5 min
- RaSP: \~10-15 minutes, depending on file size
-
How long will my session be stored?Sessions are not deleted.
-
Where is the source-code available?
https://github.com/starbeachlab/MutationExplorerServer
It is also possible to add issues if something is not working.
-
Where can I download all input files for the tutorials and examples?Next to each example you will find an info text with all the inputs and imports used to create the example. If files have been uploaded, they are also available for download via a button.
-
Which files can MutationExplorer export via the download all button?The exported zip file contains the following files:
- The pdb file that was uploaded named structure.pdb
- The pdb file for each mutation step, e.g. mut_0.pdb
- Multiple pdb files for each step storing different calculations in the B-factor:
- The absolute energy, e.g., mut_0_absE.pdb
- Energy difference to parent, e.g., mut_0_1_diffE.pdb
- Absolute hydrophobicity, e.g., mut_0_HyPh.pdb
- Hydrophobicity difference to parent, e.g., mut_0_1_diffHyPh.pdb
- Sequence conservation to parent, e.g., mut_0_1_cons.pdb
- The clustal file for the alignments for each chain and mutation step, e.g., mut_0_1_A.clw
- The RaSP predictions for each chain and mutation step, e.g., cavity_pred_mut_0_A.csv
Happy mutating!
Please cite: MutationExplorer: a webserver for mutation of proteins and 3D visualization of energetic impacts