 Version: 2014-05-21
Version: 2014-05-21
Methods
List of helical membrane proteins
MP:PD encloses all alpha helical transmembrane proteins derived from the OPM database (Lomize, Pogozheva, Joo, Mosberg, & Lomize, 2012), PDBTM (Kozma, Simon, & Tusnády, 2013), or MPstruc. MP:PD is thus a sub dataset of the wwPDB (e.g. RCSB) and lists only proteins with at least one transmembrane helix. We search all three databases for new entries when updating MP:PD. MP:PD includes entries derived from various techniques such as electron crystallography, electron microscopy, solid-state NMR, solution NMR and X-ray diffraction. Theoretical models and peripheral proteins are excluded. Internal packing densities, internal cavities and internal waters (see below) are calculated for all entries, excluding those containing only backbone atoms or low resolution structures ( 4.0 Å).
Calculation of packing densities
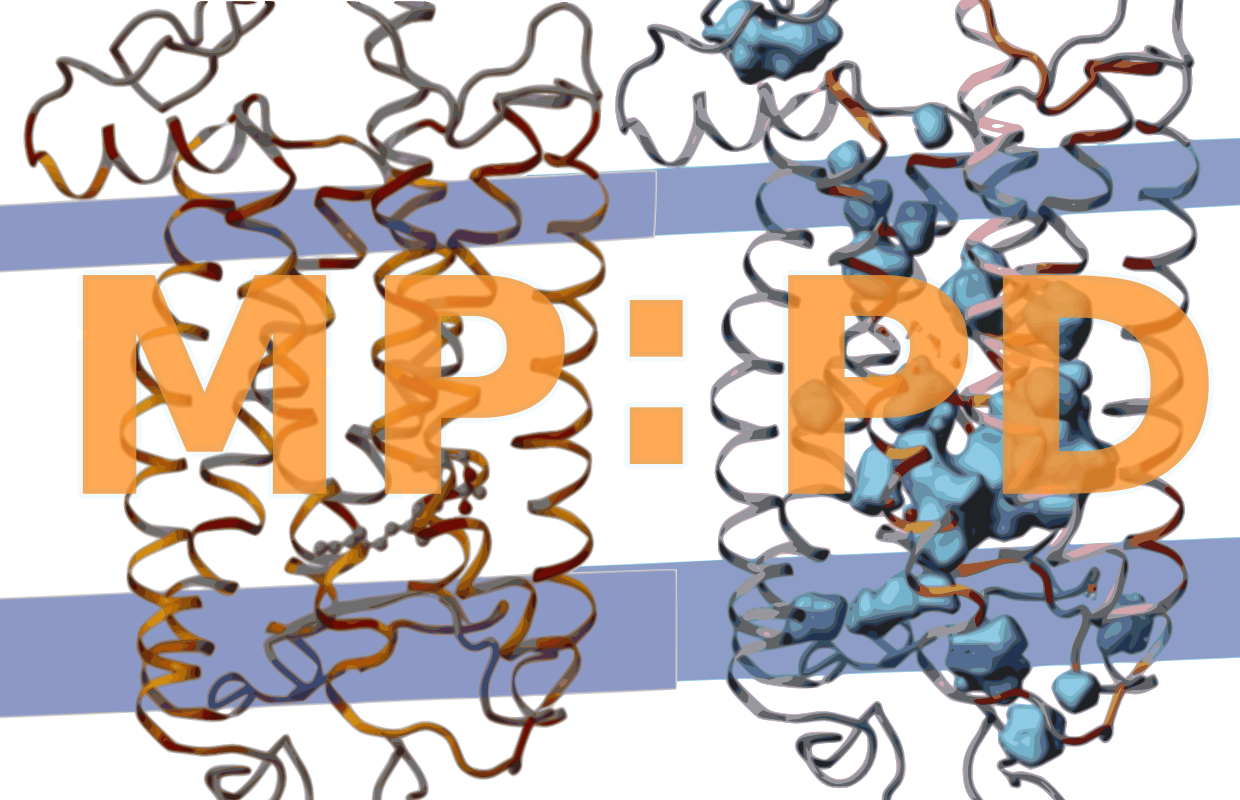
The atomic packing density quantifies the space between atoms. It allows a better approximation of van der Waals contacts and surfaces than the simple calculation of solvent excluded surfaces that does not respect packing defects enclosed therein. It uses two portions of atomic volume, the van der Waals volume V(vdW) (inside the van der Waals radius), and the solvent excluded volume V(se) (a 1.4 Å layer cushioning the vdW sphere). The Voronoi Cell algorithm (Goede, Preissner, & Frömmel, 1997) calculates, how much of the V(vdW) and V(se) is occupied by other atoms. The packing density (PD) is then calculated from the remaining volumes V(vdW) and the sum of V(vdW) and V(se) using the formula PD = V(vdW) / [V(vdW) + V(se)]. The core algorithm to calculate atomic volumes has been implemented in Delphi and an intermediate layer in Python. It calculates atomic volumes from PDB structures and produces modified PDB files from which packing densities, and tabular reports containing average volumes and densities are calculated. We used the PROTOR radius set to define atomic volumes (Tsai & Gerstein, 2002). The packing densities were calculated for the original PDB files without water and for our final structure files containing all newly assigned internal water.Calculation of internal cavities
Internal cavities are frequently found in protein domains with more than 150 amino acids (Rother, Preissner, Goede, & Frömmel, 2003).
Internal cavities are defined here as internal packing defects large enough to enclose at least a spherical probe with 1.4 Å radius which approximates the Couloumb radius of a single water molecule. ‘Internal’ means that the cavity is largely buried within the protein interior. To differentiate buried from exposed protein atoms forming internal or largely exposed cavities, respectively, we constructed a tight envelope around the protein by rolling a 2.8 Å sized spherical probe along the protein surface using the program MSMS (Sanner, Olson, & Spehner, 1996). With this definition, we also include cavities that are partially accessible to water from the bulk phase, i.e. cavities placed at invaginations of protein surfaces or within channels and pores restricted by narrow entranceways.
However, we clearly exclude wide open cavities and pockets lying at the protein surface. The accuracy of the so called solvent accessible surfaces of protein cavities can be improved if the radii of the cavity forming atoms are allowed to change depending on the polar or hydrophobic nature of the cavity (Li & Nussinov, 1998). To calculate the shape of internal cavities, we are, accordingly, using a spherical probe of 1.4 Å, the Couloumb radius of water, to calculate the surface of polar cavities (i.e. cavities including internal water, for details see next chapter) and a spherical probe of 1.7 Å, the van der Waals radius of water, to calculate the surface of hydrophobic cavities (i.e. cavities not containing internal water).
Calculation of internal water positions
To find internal water positions, not reported in the PDB entry, we used an exhaustive search of the program DOWSER (Zhang & Hermans, 1996). This program detects protein cavities and pockets and assesses their hydrophilicity in terms of energy interaction of a water molecule with the surrounding atoms. Water molecules with interaction energies below 10 kcal/mol are considered low energy waters and are selected for output. After an initial run of the 'dowserx' script we applied various runs of the 'dowser-repeat' script until no additional low energy water positions are detected. Because hetero atoms are not taken into account by DOWSER - in the absence of appropriate parameters which are not provided by that tool - we did not place internal waters in contact distance to a heteroatom. Contact distance is defined here as 3.9 Å corresponding to the cut-off distance used to define hydrogen bonds. We thus only include experimentally determined water in contact distance to hetero atoms. Water placed closer as 1.4 Å to or outside the surface (see last chapter) is defined not to be buried. We keep experimentally determined internal water only when no low energy water is detected within 2.7 Å of it, the distance of a strong water-water hydrogen bond.