Proteins are the ’machines’ of the cell, taking on manifold roles as receptors, enzymes, transporters or even molecular factories. Most biological processes involve the interaction between proteins, both in healthy systems as well as in disease. Therefore, most drugs target proteins and often impact protein-protein interactions - whether intended as desired effect or unexpected as side-effects.
Computation of the binding behaviour of proteins can complement and guide experimental studies as well as the development of protein-based therapeutics. However, calculating protein binding in atomic detail is highly time consuming and requires knowledge about the structures. While proteins are complex three-dimensional molecules, their structure, function and also binding behaviour is encoded in their amino acid sequence. In turn, the interaction between proteins can be understood to some degree from their sequence alone.
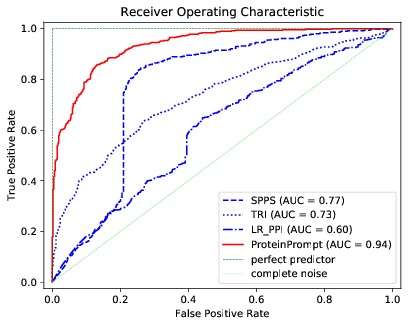
We developed ProteinPrompt, a tool based on machine learning, using the sequences of protein pairs as input to estimate their tendency to bind. The quality of its predictions outperforms any current competitor by far.
For the training of a high quality learning algorithm a comprehensive dataset needs to be collected. We exploited all available sources. Positive data was gathered from the Database of Interacting Proteins (DIP), Human Protein Reference Database (HPRD), Protein Data Bank (PDB), and the University of Kansas Proteomics Service (KUPS). Negative data was obtained from the Negatome Database [5] and the KUPS server. After collecting such a broad database, thorough curation was imperative to improve data quality. We performed several iterations of filtering and cleanup, both automated in terms of computational tools, as well as through manual inspection.
The key to ’stellar’ performance of a learning algorithm lays often in the way, the data is presented to it. Sequences can not be fed directly into learning machines, but need to be translated into descriptors that enable the learning algorithm to spot the patterns, whether a pair of sequences will bind or not. We identified auto-correlation profiles of hydrophobicity scales to best capture the binding propensity. Learning algorithms themselves are available in many implementations. Most prominent in the current rise of learning applications in biology are artificial neural networks (ANN). However, following an agnostic approach, we compared ANNs with support vector machines and several other learning methods. As a result, we found that the random forest approach leads to the cleanest separation of binders and non-binders.