Manual
Results
The top candidate loop is automatically displayed in the NGL Viewer together with the uploaded protein (model) and the user provided cryo-EM density map.
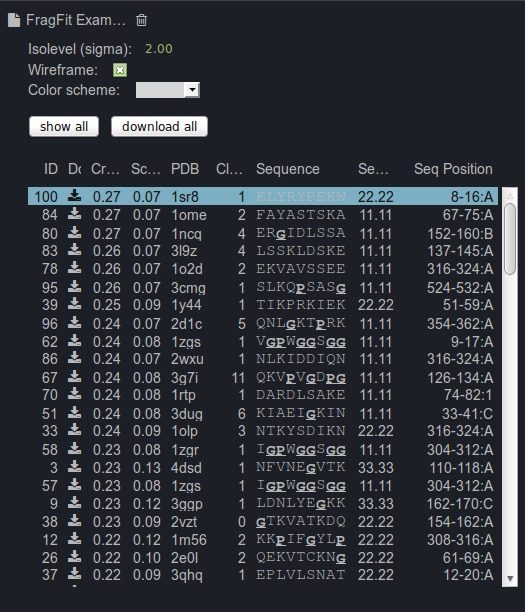
Other loops can be selected from the list of up tp 100 candidates by visualization or by using the information
provided in the table at the right panel for guidance. This table includes the following information for
each candidate loop: an ID, a correlation score, a score describing the steric fit of the segment, a clash score, the PDB ID and the position of the fragment in the
template protein and a comparison of the user-defined input sequence with the template sequence (sequence similarity).
Sequences that are underlined contain prolines or glycines.
By selecting a result, the loop is displayed together with the uploaded protein. The complete list of loop candidates can be displayed
simultaneously to visualize the conformational space of the loop. The loop can be colored accordingly to its score, correlation score, sequence identity or
clash score by selecting the corresponding color scheme from the dropdown menu. Additionally, the representation of the structure and
the loop (collapsed underneath the result table) can be changed as explained separatly in the documentation of the NGL Viewer.
The completed structure (initial model plus selected loop) can be downloaded by clicking the download symbol. Alternatively, the complete
list of loops can be downloaded for further analyses. The loops contain sidechains, which are not minimized and also not used during density fit but can be utilized during visual comparison and selection.
|
 |
Example sets
Cryo-EM structure of the human gamma-secretase complex at 3.4 angstrom resolution.
-
Download the map from EMDB: EMD-3061 and upload it to the server via the form
-
Downlaod PDB file: 5a63 and upload it to the server via the form
-
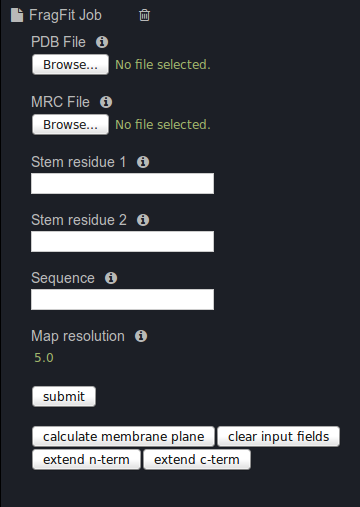
Type in the stem residues:
Stem residue 1: 473:A
Stem residue 2: 482:A
-
Type in the missing sequence: EDLNFVT
-
Add the map resolution: 3.4
-
Press start.
-
Visualize results.
Download further example data sets here:
5oby
6bo8
A detailed youtube tutorial can also be found
here.
Validation
A validation can be done by using
MolProbity. For the given example sets we found that Fragfit could improve the MolProbity Score. It can happen that the downloaded structure from Fragfit can not be uploaded to MolProbity, if that happens do the following: Copy the atoms of the loop from the Fragfit result into the original pdb and delete the original loop, then upload this file.